LIANA Algorithm Principles and Mathematical Framework
Zaoqu Liu
Fork Maintainerliuzaoqu@163.com
Daniel Dimitrov
Saezlab, Heidelberg University2026-01-23
Source:vignettes/algorithms.Rmd
algorithms.RmdOverview
LIANA (LIgand-receptor ANalysis frAmework) integrates multiple cell-cell communication (CCC) inference methods. This vignette provides a comprehensive mathematical description of each scoring algorithm implemented in LIANA.
Cell-Cell Communication Inference
The fundamental premise of CCC inference from scRNA-seq data is that the expression levels of ligand genes in “sender” cells and receptor genes in “receiver” cells can indicate potential intercellular signaling.
For a ligand expressed in cell type and receptor expressed in cell type , we seek to quantify:
- Expression Magnitude: How strongly are and expressed?
- Expression Specificity: How specific is this - interaction to the - cell type pair?
Scoring Methods
1. SingleCellSignalR (LRScore)
2. NATMI (Edge Specificity)
Mathematical Formulation
NATMI calculates specificity-weighted edge scores:
Where: - = sum of ligand expression across all cell types - = sum of receptor expression across all cell types
3. Connectome (Scaled Weights)
4. CellPhoneDB (Permutation-based P-values)
Consensus Ranking
Complex Handling
For heteromeric complexes (e.g., interacting with ), LIANA applies a policy function:
Available policies: - mean0: Mean, but returns 0 if any
subunit is absent - min: Minimum subunit score (limiting
factor) - geometric_mean: Geometric mean of subunits
Visualization of Score Distributions
library(liana)
library(ggplot2)
library(dplyr)
library(tidyr)
# Load example data
liana_path <- system.file(package = "liana")
testdata <- readRDS(file.path(liana_path, "testdata", "input", "testdata.rds"))
# Run LIANA
liana_res <- liana_wrap(testdata,
method = c("natmi", "sca", "connectome", "logfc"),
resource = "Consensus")
# Aggregate results
liana_aggr <- liana_aggregate(liana_res)
# Visualize score distributions
liana_aggr %>%
select(ends_with("rank")) %>%
pivot_longer(everything(), names_to = "Method", values_to = "Rank") %>%
mutate(Method = gsub("\\.rank", "", Method)) %>%
ggplot(aes(x = Rank, fill = Method)) +
geom_density(alpha = 0.5) +
theme_minimal(base_size = 12) +
labs(title = "Distribution of Ranks Across Methods",
x = "Rank", y = "Density") +
theme(legend.position = "bottom")
Score Correlation Heatmap
# Calculate correlations between method scores
score_cols <- liana_aggr %>%
select(ends_with("rank")) %>%
cor(use = "complete.obs")
# Plot correlation heatmap
score_cols %>%
as.data.frame() %>%
tibble::rownames_to_column("Method1") %>%
pivot_longer(-Method1, names_to = "Method2", values_to = "Correlation") %>%
mutate(Method1 = gsub("\\.rank", "", Method1),
Method2 = gsub("\\.rank", "", Method2)) %>%
ggplot(aes(x = Method1, y = Method2, fill = Correlation)) +
geom_tile() +
geom_text(aes(label = round(Correlation, 2)), color = "white", size = 5) +
scale_fill_gradient2(low = "blue", mid = "white", high = "red", midpoint = 0.5) +
theme_minimal(base_size = 12) +
labs(title = "Rank Correlation Between Methods",
x = "", y = "") +
theme(axis.text.x = element_text(angle = 45, hjust = 1))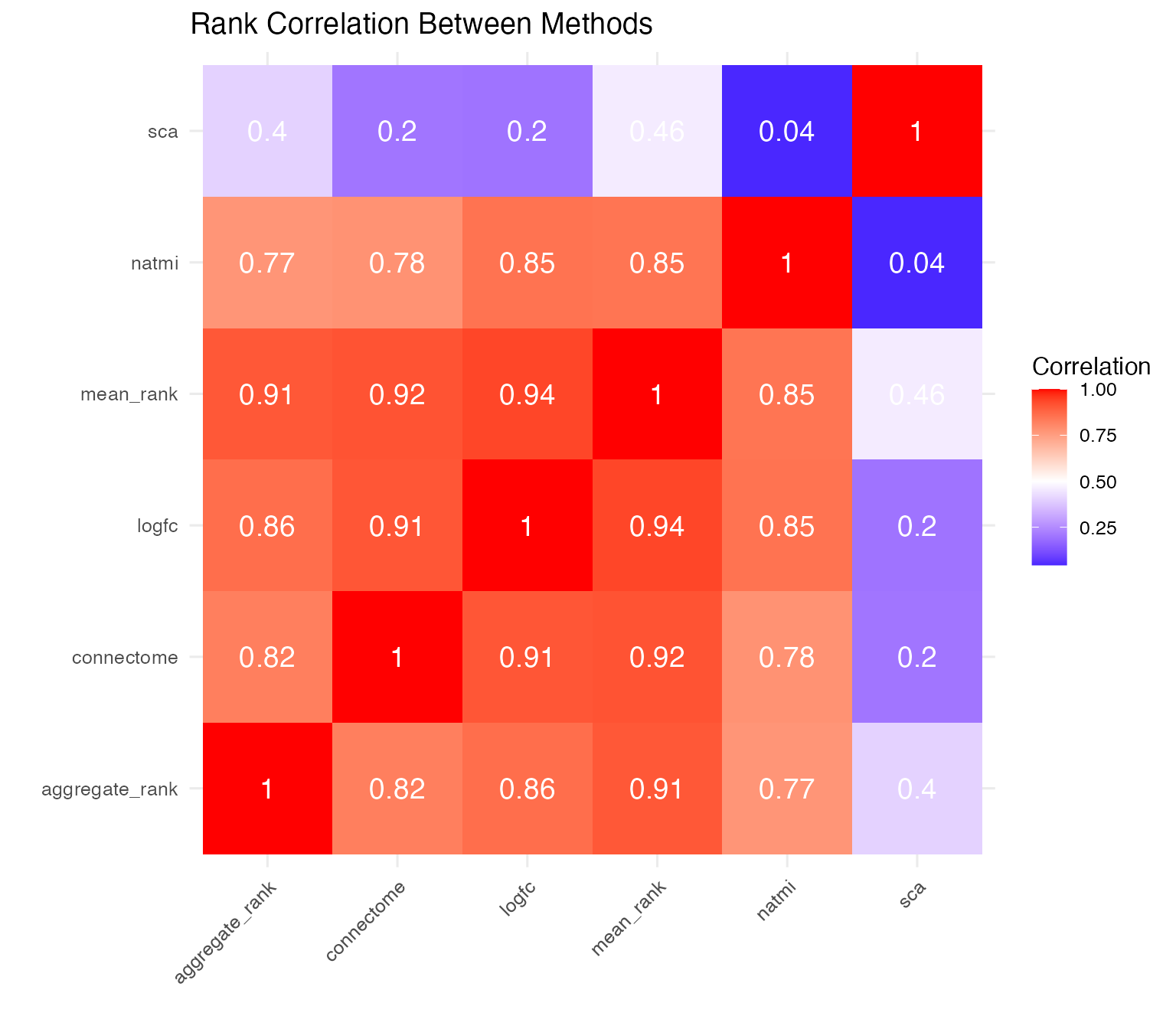
Summary Table
| Method | Score Type | Range | Interpretation | Best For |
|---|---|---|---|---|
| SingleCellSignalR | Magnitude | [0,1] | Higher = stronger | Cross-dataset comparison |
| NATMI | Specificity | [0,1] | Higher = more specific | Cell-type specific interactions |
| Connectome | Z-score | Unbounded | Higher = enriched | Relative expression |
| CellPhoneDB | P-value | [0,1] | Lower = significant | Statistical validation |
| LogFC | Fold-change | Unbounded | Higher = enriched | Differential interactions |
| CytoTalk | Crosstalk | [0,1] | Higher = stronger | Autocrine/paracrine distinction |
References
Dimitrov D, et al. (2022). Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nature Communications.
Cabello-Aguilar S, et al. (2020). SingleCellSignalR: inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Research.
Hou R, et al. (2020). Predicting cell-to-cell communication networks using NATMI. Nature Communications.
Raredon MSB, et al. (2019). Single-cell connectomic analysis of adult mammalian lungs. Science Advances.
Efremova M, et al. (2020). CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nature Protocols.
Session Information
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] tidyr_1.3.2 dplyr_1.1.4 ggplot2_4.0.1 liana_0.1.14.9000
#> [5] BiocStyle_2.34.0
#>
#> loaded via a namespace (and not attached):
#> [1] spatstat.sparse_3.1-0 fs_1.6.6
#> [3] matrixStats_1.5.0 lubridate_1.9.4
#> [5] httr_1.4.7 RColorBrewer_1.1-3
#> [7] doParallel_1.0.17 tools_4.4.0
#> [9] sctransform_0.4.3 backports_1.5.0
#> [11] R6_2.6.1 uwot_0.2.4
#> [13] lazyeval_0.2.2 GetoptLong_1.1.0
#> [15] withr_3.0.2 sp_2.2-0
#> [17] gridExtra_2.3 prettyunits_1.2.0
#> [19] progressr_0.18.0 cli_3.6.5
#> [21] Biobase_2.66.0 textshaping_1.0.4
#> [23] spatstat.explore_3.6-0 labeling_0.4.3
#> [25] sass_0.4.10 Seurat_4.4.0
#> [27] spatstat.data_3.1-9 S7_0.2.1
#> [29] readr_2.1.6 ggridges_0.5.7
#> [31] pbapply_1.7-4 pkgdown_2.1.3
#> [33] systemfonts_1.3.1 R.utils_2.13.0
#> [35] scater_1.34.1 dichromat_2.0-0.1
#> [37] parallelly_1.46.1 sessioninfo_1.2.3
#> [39] limma_3.62.2 readxl_1.4.5
#> [41] RSQLite_2.4.5 generics_0.1.4
#> [43] shape_1.4.6.1 spatstat.random_3.4-3
#> [45] ica_1.0-3 zip_2.3.3
#> [47] Matrix_1.7-4 ggbeeswarm_0.7.3
#> [49] S4Vectors_0.44.0 logger_0.4.1
#> [51] abind_1.4-8 R.methodsS3_1.8.2
#> [53] lifecycle_1.0.5 yaml_2.3.12
#> [55] edgeR_4.4.2 SummarizedExperiment_1.36.0
#> [57] SparseArray_1.6.2 Rtsne_0.17
#> [59] grid_4.4.0 blob_1.2.4
#> [61] promises_1.5.0 dqrng_0.4.1
#> [63] crayon_1.5.3 dir.expiry_1.14.0
#> [65] miniUI_0.1.2 lattice_0.22-7
#> [67] beachmat_2.22.0 cowplot_1.2.0
#> [69] chromote_0.5.1 pillar_1.11.1
#> [71] knitr_1.51 ComplexHeatmap_2.22.0
#> [73] metapod_1.14.0 GenomicRanges_1.58.0
#> [75] tcltk_4.4.0 rjson_0.2.23
#> [77] future.apply_1.20.1 codetools_0.2-20
#> [79] leiden_0.4.3.1 glue_1.8.0
#> [81] spatstat.univar_3.1-6 data.table_1.18.0
#> [83] vctrs_0.7.0 png_0.1-8
#> [85] cellranger_1.1.0 gtable_0.3.6
#> [87] cachem_1.1.0 OmnipathR_3.19.1
#> [89] xfun_0.56 S4Arrays_1.6.0
#> [91] mime_0.13 survival_3.8-3
#> [93] SingleCellExperiment_1.28.1 iterators_1.0.14
#> [95] statmod_1.5.1 bluster_1.16.0
#> [97] fitdistrplus_1.2-4 ROCR_1.0-11
#> [99] nlme_3.1-168 bit64_4.6.0-1
#> [101] progress_1.2.3 filelock_1.0.3
#> [103] RcppAnnoy_0.0.23 GenomeInfoDb_1.42.3
#> [105] bslib_0.9.0 irlba_2.3.5.1
#> [107] vipor_0.4.7 KernSmooth_2.23-26
#> [109] otel_0.2.0 colorspace_2.1-2
#> [111] BiocGenerics_0.52.0 DBI_1.2.3
#> [113] tidyselect_1.2.1 processx_3.8.6
#> [115] bit_4.6.0 compiler_4.4.0
#> [117] curl_7.0.0 rvest_1.0.5
#> [119] httr2_1.2.2 BiocNeighbors_2.0.1
#> [121] xml2_1.5.2 desc_1.4.3
#> [123] DelayedArray_0.32.0 plotly_4.11.0
#> [125] bookdown_0.44 checkmate_2.3.3
#> [127] scales_1.4.0 lmtest_0.9-40
#> [129] rappdirs_0.3.4 goftest_1.2-3
#> [131] stringr_1.6.0 digest_0.6.39
#> [133] spatstat.utils_3.2-1 rmarkdown_2.30
#> [135] basilisk_1.23.0 XVector_0.46.0
#> [137] htmltools_0.5.9 pkgconfig_2.0.3
#> [139] sparseMatrixStats_1.18.0 MatrixGenerics_1.18.1
#> [141] fastmap_1.2.0 rlang_1.1.7
#> [143] GlobalOptions_0.1.3 htmlwidgets_1.6.4
#> [145] UCSC.utils_1.2.0 shiny_1.12.1
#> [147] farver_2.1.2 jquerylib_0.1.4
#> [149] zoo_1.8-15 jsonlite_2.0.0
#> [151] BiocParallel_1.40.2 R.oo_1.27.1
#> [153] BiocSingular_1.22.0 magrittr_2.0.4
#> [155] scuttle_1.16.0 GenomeInfoDbData_1.2.13
#> [157] patchwork_1.3.2 Rcpp_1.1.1
#> [159] viridis_0.6.5 reticulate_1.44.1
#> [161] stringi_1.8.7 zlibbioc_1.52.0
#> [163] MASS_7.3-65 plyr_1.8.9
#> [165] parallel_4.4.0 listenv_0.10.0
#> [167] ggrepel_0.9.6 deldir_2.0-4
#> [169] splines_4.4.0 tensor_1.5.1
#> [171] hms_1.1.4 circlize_0.4.17
#> [173] locfit_1.5-9.12 ps_1.9.1
#> [175] igraph_2.2.1 spatstat.geom_3.6-1
#> [177] reshape2_1.4.5 stats4_4.4.0
#> [179] ScaledMatrix_1.14.0 XML_3.99-0.20
#> [181] evaluate_1.0.5 SeuratObject_4.1.4
#> [183] scran_1.34.0 BiocManager_1.30.27
#> [185] tzdb_0.5.0 foreach_1.5.2
#> [187] httpuv_1.6.16 polyclip_1.10-7
#> [189] RANN_2.6.2 purrr_1.2.1
#> [191] future_1.69.0 clue_0.3-66
#> [193] scattermore_1.2 rsvd_1.0.5
#> [195] xtable_1.8-4 later_1.4.5
#> [197] viridisLite_0.4.2 ragg_1.5.0
#> [199] tibble_3.3.1 websocket_1.4.4
#> [201] beeswarm_0.4.0 memoise_2.0.1
#> [203] IRanges_2.40.1 cluster_2.1.8.1
#> [205] timechange_0.3.0 globals_0.18.0