LIANA Visualization Guide
Zaoqu Liu
Fork Maintainerliuzaoqu@163.com
2026-01-23
Source:vignettes/visualization.Rmd
visualization.RmdOverview
This vignette demonstrates various visualization options available in LIANA for exploring cell-cell communication results.
Run LIANA Analysis
# Run analysis
liana_res <- liana_wrap(testdata,
method = c("natmi", "connectome", "logfc", "sca"),
resource = "Consensus"
)
# Aggregate results
liana_aggr <- liana_aggregate(liana_res)1. Dotplot Visualizations
Basic Dotplot
The dotplot is the primary visualization for LIANA results, showing: - Color: Expression magnitude (LRscore) - Size: Interaction specificity (edge_specificity)
liana_aggr %>%
liana_dotplot(ntop = 20)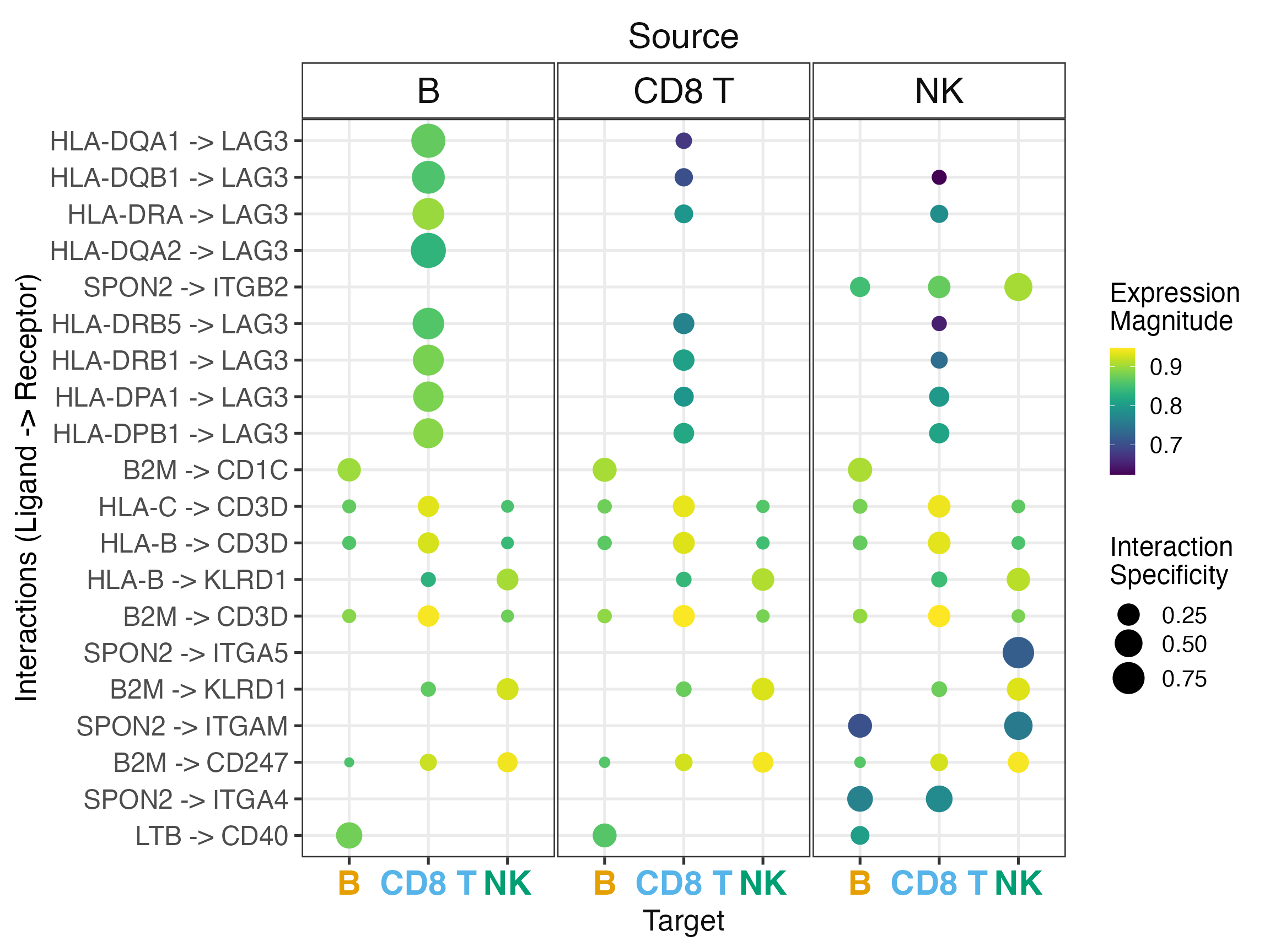
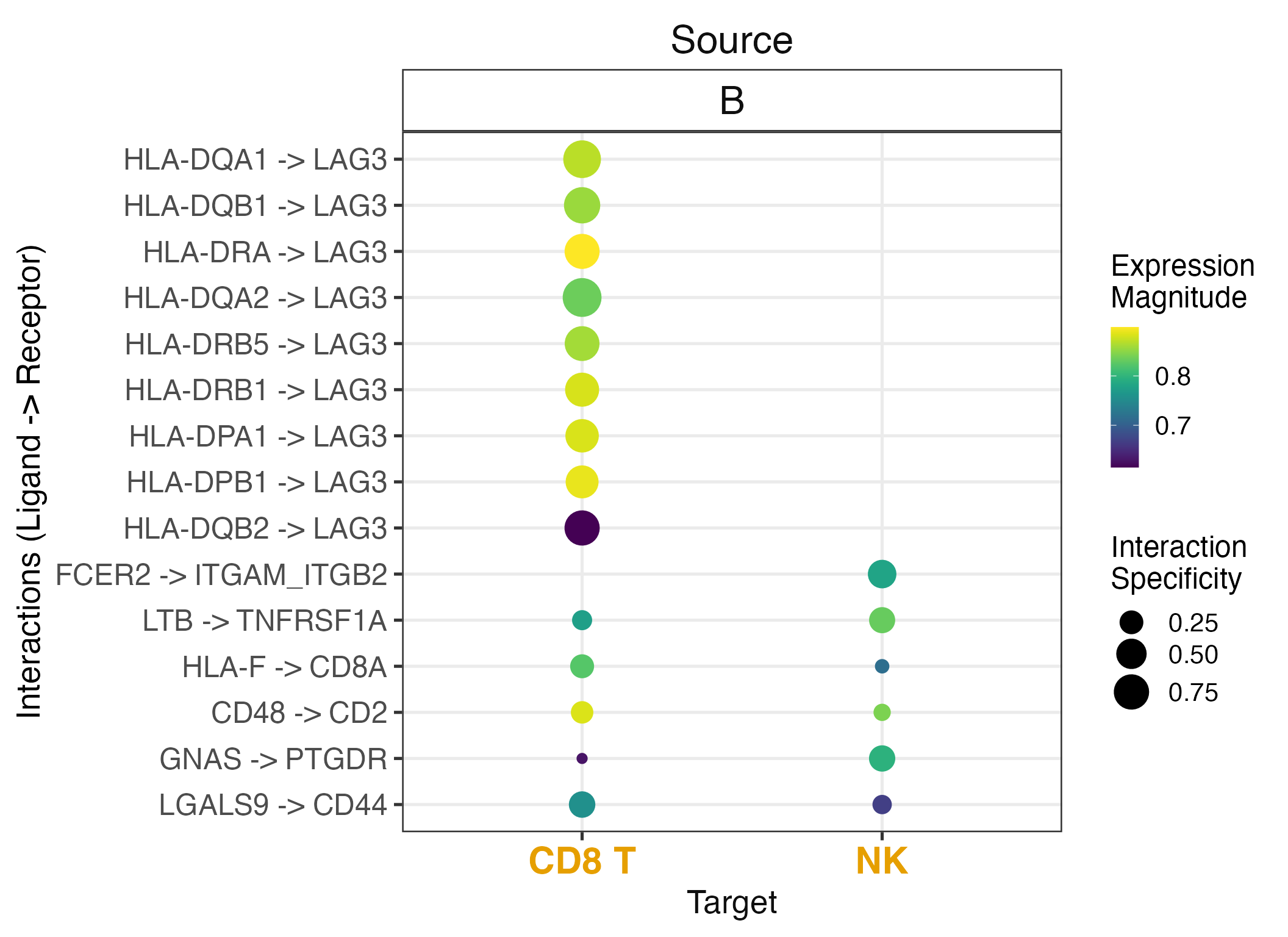
Filtered Dotplot
# Filter by source and target cell types
liana_aggr %>%
liana_dotplot(
source_groups = c("B"),
target_groups = c("NK", "CD8 T"),
ntop = 15
)
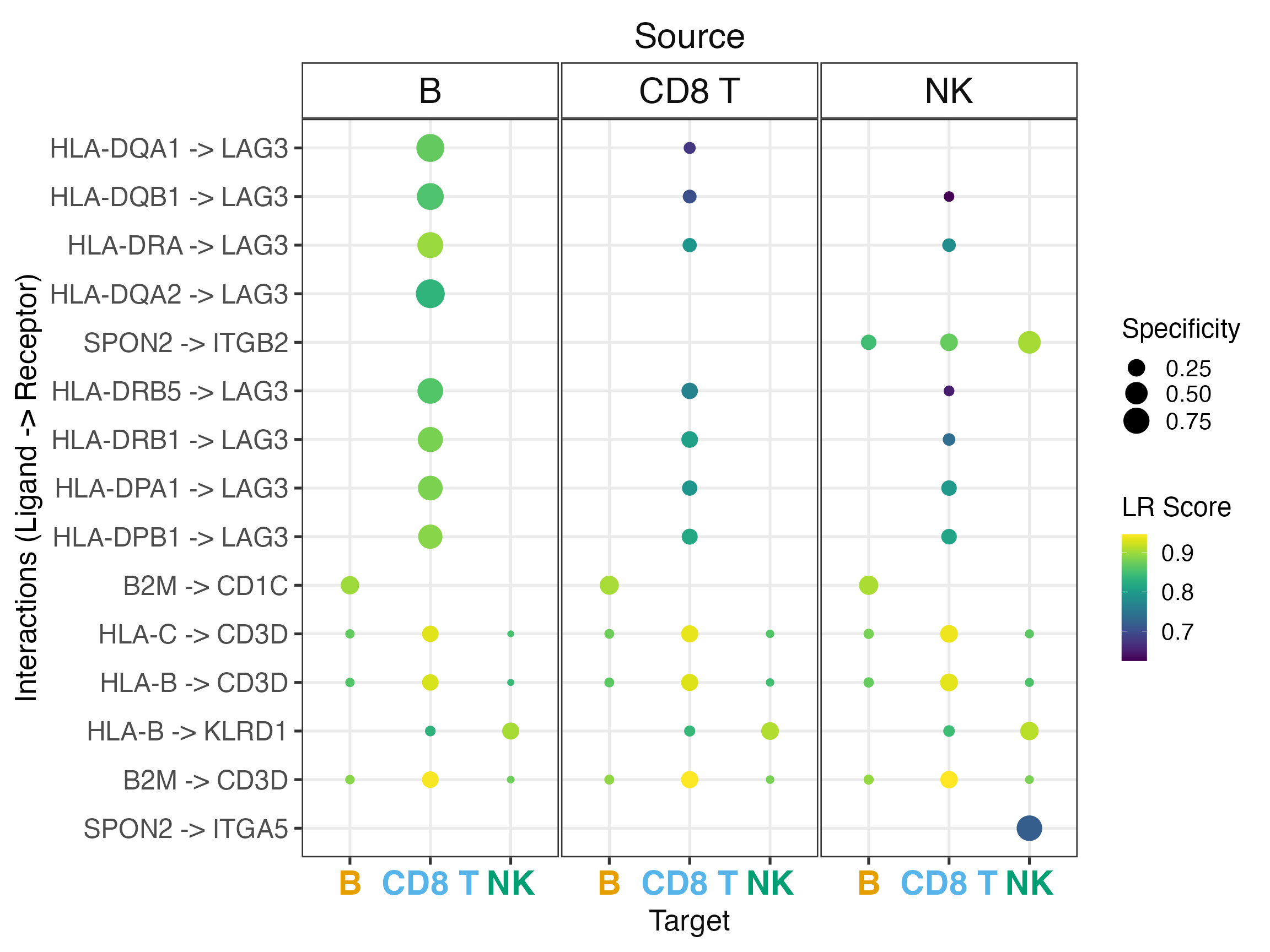
Custom Dotplot Parameters
# Use different metrics and adjust size
liana_aggr %>%
liana_dotplot(
ntop = 15,
specificity = "natmi.edge_specificity",
magnitude = "sca.LRscore",
size_range = c(1, 8),
size.label = "Specificity",
colour.label = "LR Score"
)
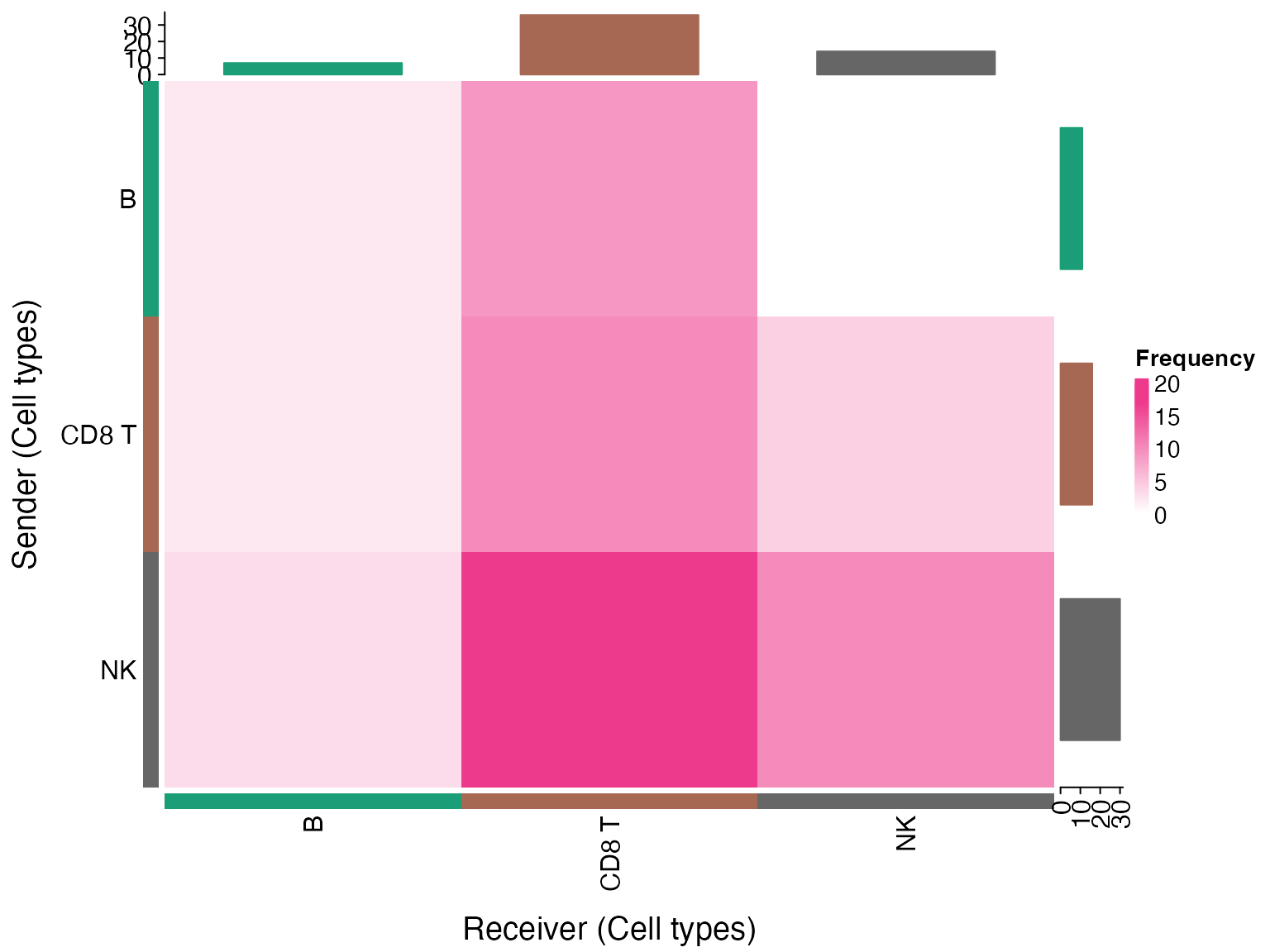
3. Chord Diagrams
Chord diagrams provide a circular visualization of cell-cell communication.
# Check if circlize is available
if(requireNamespace("circlize", quietly = TRUE) && nrow(liana_sig) > 0) {
chord_freq(liana_sig)
}
Subset Chord Diagram
if(requireNamespace("circlize", quietly = TRUE) && nrow(liana_sig) > 0) {
chord_freq(liana_sig,
source_groups = c("CD8 T", "NK"),
target_groups = c("CD8 T", "NK", "B"),
cex = 1.2
)
}
4. Custom ggplot2 Visualizations
Method Comparison Plot
# Compare rankings across methods
liana_aggr %>%
head(50) %>%
select(ligand.complex, receptor.complex, source, target,
ends_with(".rank")) %>%
pivot_longer(cols = ends_with(".rank"),
names_to = "method",
values_to = "rank") %>%
mutate(method = gsub("\\.rank", "", method)) %>%
mutate(interaction = paste(ligand.complex, receptor.complex, sep = " -> ")) %>%
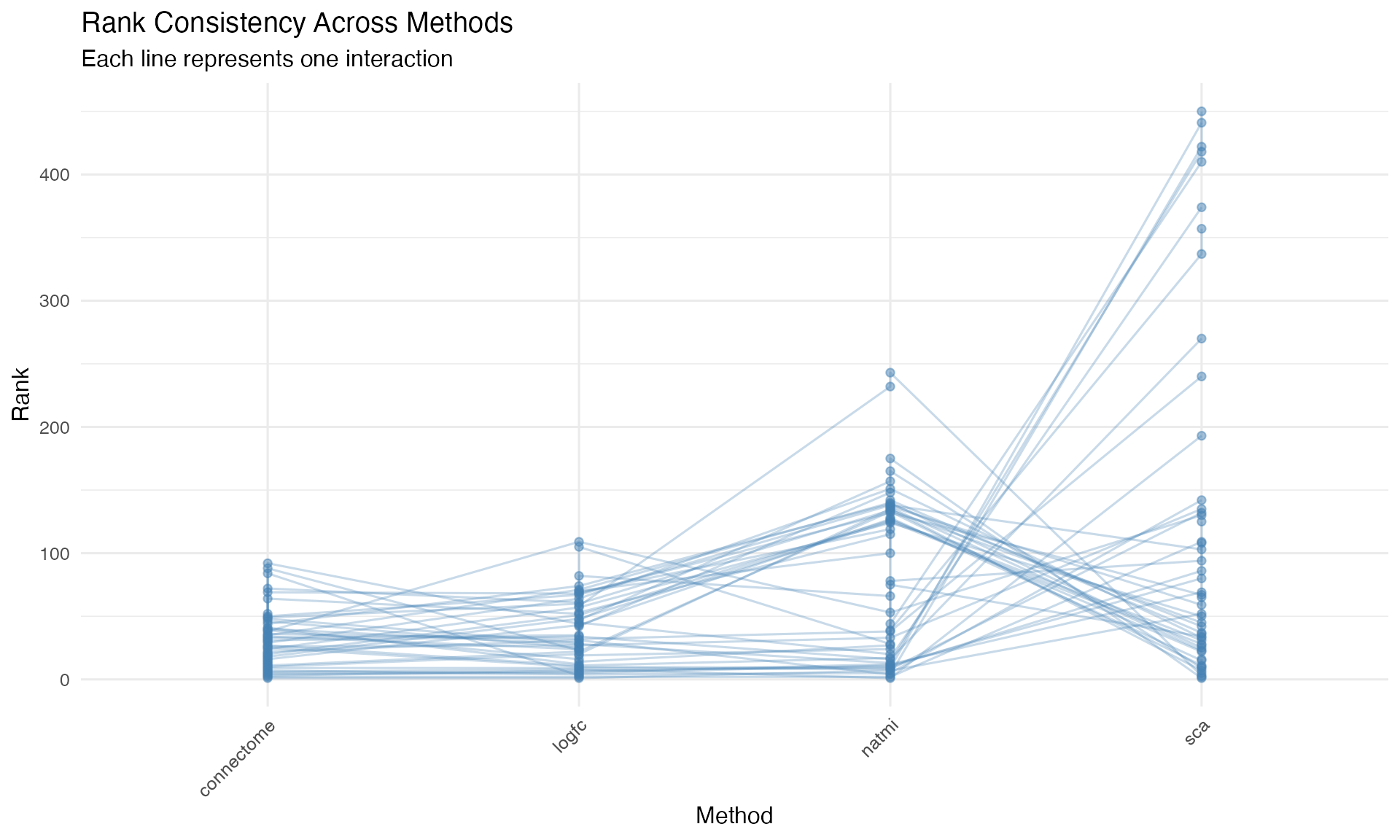
ggplot(aes(x = method, y = rank, group = interaction)) +
geom_line(alpha = 0.3, color = "steelblue") +
geom_point(alpha = 0.5, color = "steelblue") +
theme_minimal(base_size = 12) +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
labs(
title = "Rank Consistency Across Methods",
subtitle = "Each line represents one interaction",
x = "Method",
y = "Rank"
)
Score Distribution Plot
# Score distributions
liana_aggr %>%
select(sca.LRscore, natmi.edge_specificity,
connectome.weight_sc, logfc.logfc_comb) %>%
pivot_longer(everything(), names_to = "Score", values_to = "Value") %>%
ggplot(aes(x = Value, fill = Score)) +
geom_density(alpha = 0.5) +
facet_wrap(~Score, scales = "free") +
theme_minimal(base_size = 11) +
theme(legend.position = "none") +
labs(title = "Distribution of LIANA Scores")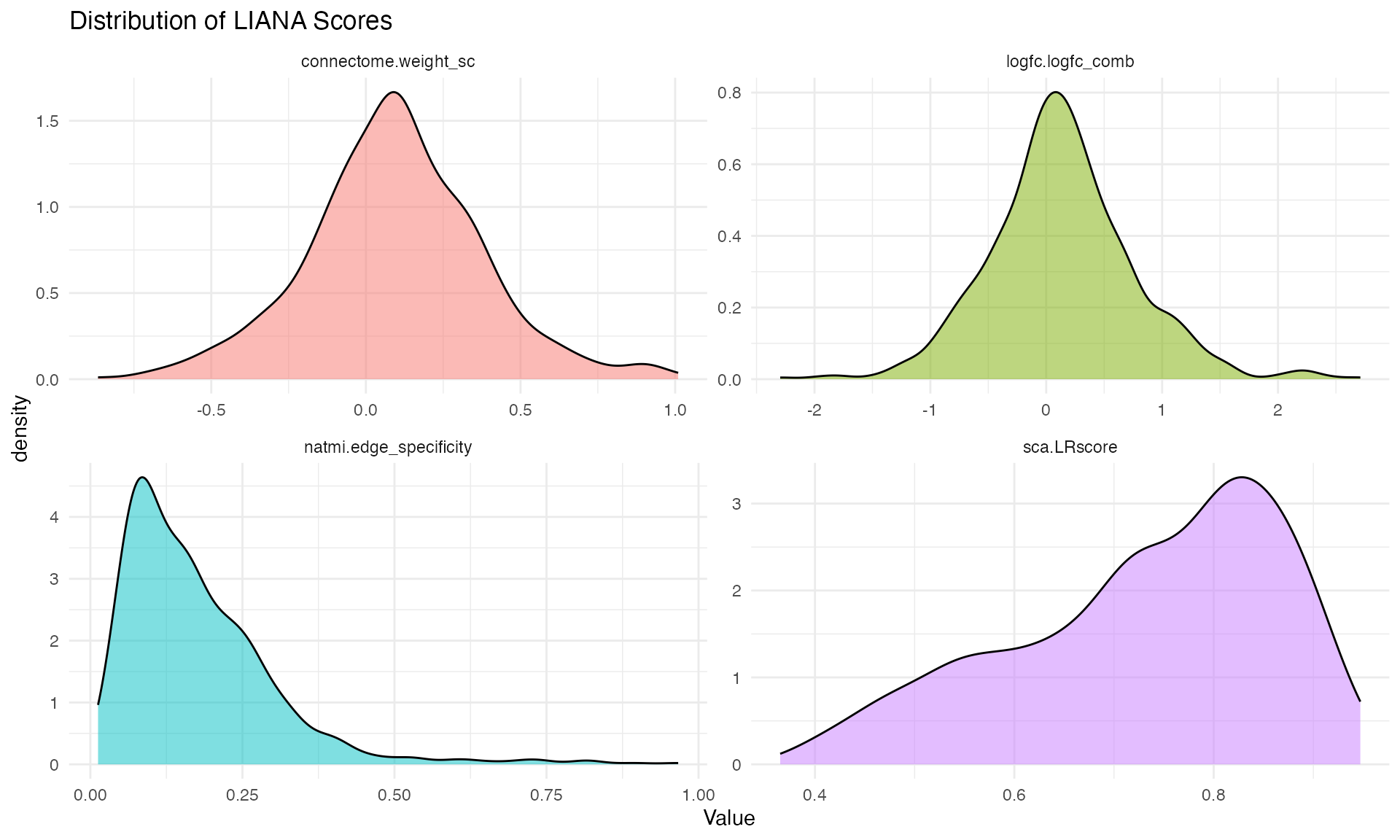
Top Interactions Bar Plot
liana_aggr %>%
head(15) %>%
mutate(interaction = paste(ligand.complex, "→", receptor.complex)) %>%
mutate(cell_pair = paste(source, "→", target)) %>%
ggplot(aes(x = reorder(interaction, -aggregate_rank),
y = -log10(aggregate_rank + 1e-10),
fill = cell_pair)) +
geom_col() +
coord_flip() +
theme_minimal(base_size = 11) +
labs(
title = "Top 15 Cell-Cell Interactions",
x = "Interaction (Ligand → Receptor)",
y = "-log10(Aggregate Rank)",
fill = "Cell Pair"
) +
theme(legend.position = "right")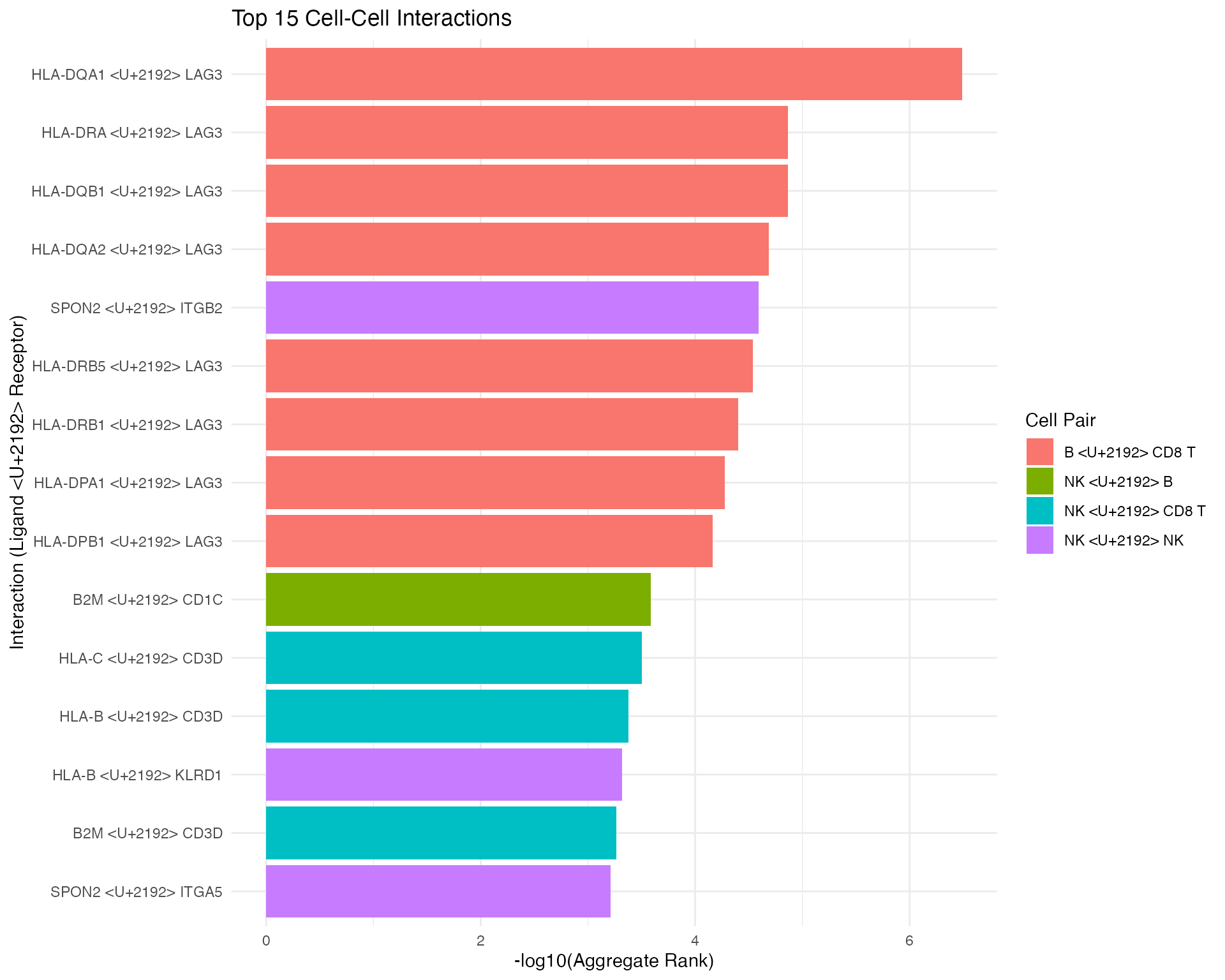
5. Network Visualization
Build Interaction Network
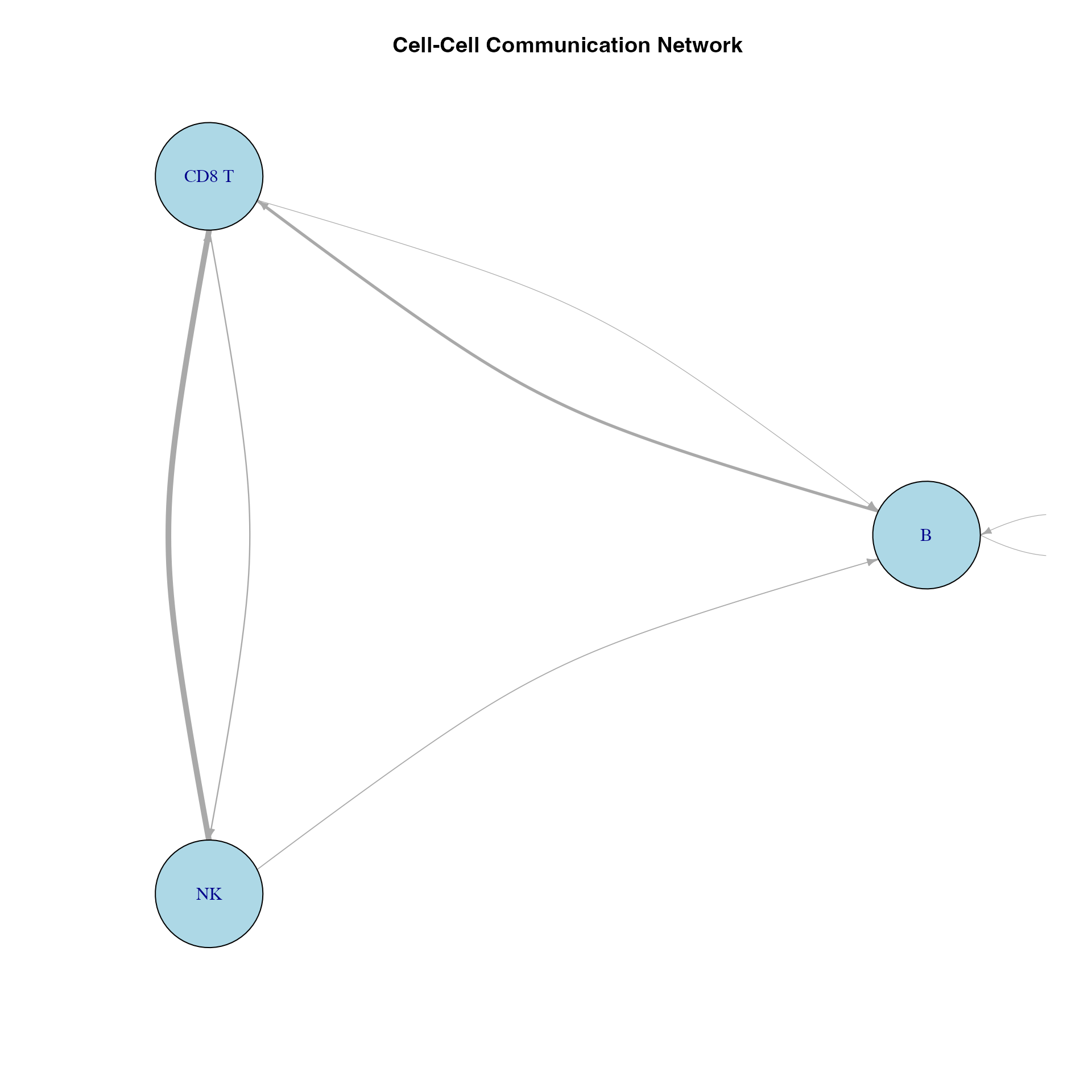
if(requireNamespace("igraph", quietly = TRUE) && nrow(liana_sig) > 0) {
library(igraph)
# Create edge list
edges <- liana_sig %>%
group_by(source, target) %>%
summarise(weight = n(), .groups = "drop")
# Create graph
g <- graph_from_data_frame(edges, directed = TRUE)
E(g)$width <- E(g)$weight / max(E(g)$weight) * 5
# Plot
plot(g,
vertex.size = 30,
vertex.label.cex = 1,
vertex.color = "lightblue",
edge.arrow.size = 0.5,
edge.curved = 0.2,
layout = layout_in_circle,
main = "Cell-Cell Communication Network"
)
}
6. Publication-Ready Figures
Combined Figure
library(patchwork)
# Create individual plots
p1 <- liana_aggr %>%
head(10) %>%
mutate(interaction = paste(ligand.complex, "→", receptor.complex)) %>%
ggplot(aes(x = reorder(interaction, -aggregate_rank),
y = sca.LRscore, fill = source)) +
geom_col() +
coord_flip() +
theme_minimal() +
labs(title = "A. Top Interactions by LRscore", x = "", y = "LR Score")
p2 <- liana_aggr %>%
group_by(source, target) %>%
summarise(n_int = sum(aggregate_rank <= 0.05), .groups = "drop") %>%
ggplot(aes(x = source, y = target, fill = n_int)) +
geom_tile() +
scale_fill_viridis_c() +
theme_minimal() +
labs(title = "B. Significant Interactions per Cell Pair",
fill = "Count", x = "Source", y = "Target")
p3 <- liana_aggr %>%
ggplot(aes(x = sca.LRscore, y = natmi.edge_specificity,
color = aggregate_rank <= 0.01)) +
geom_point(alpha = 0.5) +
scale_color_manual(values = c("gray70", "red"),
labels = c("NS", "Significant")) +
theme_minimal() +
labs(title = "C. Magnitude vs Specificity",
x = "LR Score (Magnitude)",
y = "Edge Specificity",
color = "")
# Combine
(p1 | p2) / p3 +
plot_annotation(title = "LIANA Cell-Cell Communication Analysis")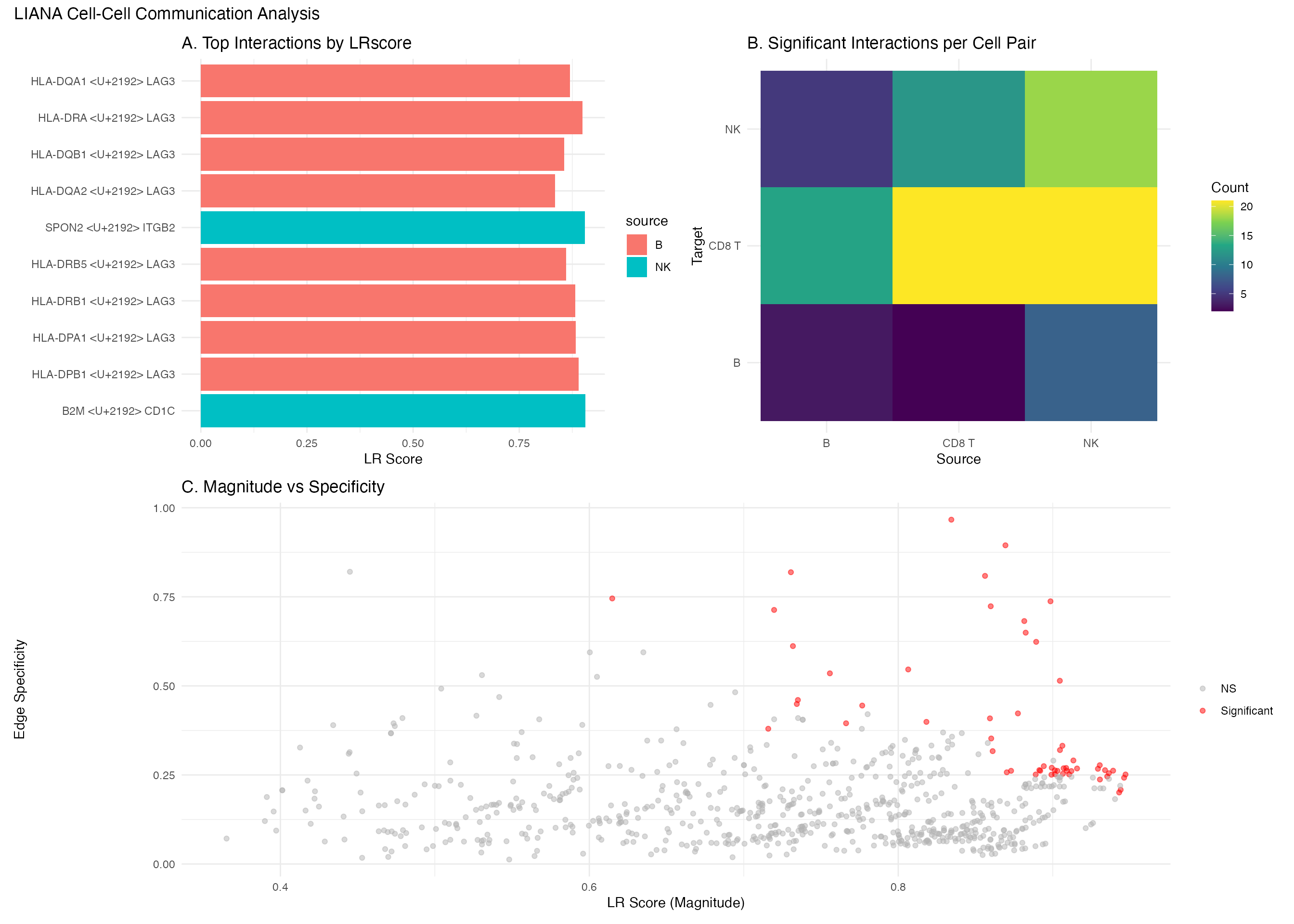
Tips for Effective Visualization
- Filter First: Always filter to significant interactions before plotting
-
Choose Appropriate Metrics:
- Magnitude for expression strength
- Specificity for cell-type exclusivity
-
Consider Your Audience:
- Dotplots for detailed inspection
- Heatmaps for overview
- Chord diagrams for presentations
- Export High Resolution:
ggsave("liana_figure.pdf", width = 12, height = 8, dpi = 300)Session Information
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] patchwork_1.3.2 igraph_2.2.1 tidyr_1.3.2 ggplot2_4.0.1
#> [5] dplyr_1.1.4 liana_0.1.14.9000 BiocStyle_2.34.0
#>
#> loaded via a namespace (and not attached):
#> [1] spatstat.sparse_3.1-0 fs_1.6.6
#> [3] matrixStats_1.5.0 lubridate_1.9.4
#> [5] httr_1.4.7 RColorBrewer_1.1-3
#> [7] doParallel_1.0.17 tools_4.4.0
#> [9] sctransform_0.4.3 backports_1.5.0
#> [11] R6_2.6.1 uwot_0.2.4
#> [13] lazyeval_0.2.2 GetoptLong_1.1.0
#> [15] withr_3.0.2 sp_2.2-0
#> [17] gridExtra_2.3 prettyunits_1.2.0
#> [19] progressr_0.18.0 cli_3.6.5
#> [21] Biobase_2.66.0 textshaping_1.0.4
#> [23] Cairo_1.7-0 spatstat.explore_3.6-0
#> [25] labeling_0.4.3 sass_0.4.10
#> [27] Seurat_4.4.0 spatstat.data_3.1-9
#> [29] S7_0.2.1 readr_2.1.6
#> [31] ggridges_0.5.7 pbapply_1.7-4
#> [33] pkgdown_2.1.3 systemfonts_1.3.1
#> [35] R.utils_2.13.0 scater_1.34.1
#> [37] dichromat_2.0-0.1 parallelly_1.46.1
#> [39] sessioninfo_1.2.3 limma_3.62.2
#> [41] readxl_1.4.5 RSQLite_2.4.5
#> [43] generics_0.1.4 shape_1.4.6.1
#> [45] spatstat.random_3.4-3 ica_1.0-3
#> [47] zip_2.3.3 Matrix_1.7-4
#> [49] ggbeeswarm_0.7.3 S4Vectors_0.44.0
#> [51] logger_0.4.1 abind_1.4-8
#> [53] R.methodsS3_1.8.2 lifecycle_1.0.5
#> [55] yaml_2.3.12 edgeR_4.4.2
#> [57] SummarizedExperiment_1.36.0 SparseArray_1.6.2
#> [59] Rtsne_0.17 grid_4.4.0
#> [61] blob_1.2.4 promises_1.5.0
#> [63] dqrng_0.4.1 crayon_1.5.3
#> [65] dir.expiry_1.14.0 miniUI_0.1.2
#> [67] lattice_0.22-7 beachmat_2.22.0
#> [69] cowplot_1.2.0 chromote_0.5.1
#> [71] magick_2.8.7 pillar_1.11.1
#> [73] knitr_1.51 ComplexHeatmap_2.22.0
#> [75] metapod_1.14.0 GenomicRanges_1.58.0
#> [77] tcltk_4.4.0 rjson_0.2.23
#> [79] future.apply_1.20.1 codetools_0.2-20
#> [81] leiden_0.4.3.1 glue_1.8.0
#> [83] spatstat.univar_3.1-6 data.table_1.18.0
#> [85] vctrs_0.7.0 png_0.1-8
#> [87] cellranger_1.1.0 gtable_0.3.6
#> [89] cachem_1.1.0 OmnipathR_3.19.1
#> [91] xfun_0.56 S4Arrays_1.6.0
#> [93] mime_0.13 survival_3.8-3
#> [95] SingleCellExperiment_1.28.1 iterators_1.0.14
#> [97] statmod_1.5.1 bluster_1.16.0
#> [99] fitdistrplus_1.2-4 ROCR_1.0-11
#> [101] nlme_3.1-168 bit64_4.6.0-1
#> [103] progress_1.2.3 filelock_1.0.3
#> [105] RcppAnnoy_0.0.23 GenomeInfoDb_1.42.3
#> [107] bslib_0.9.0 irlba_2.3.5.1
#> [109] vipor_0.4.7 KernSmooth_2.23-26
#> [111] otel_0.2.0 colorspace_2.1-2
#> [113] BiocGenerics_0.52.0 DBI_1.2.3
#> [115] tidyselect_1.2.1 processx_3.8.6
#> [117] bit_4.6.0 compiler_4.4.0
#> [119] curl_7.0.0 rvest_1.0.5
#> [121] httr2_1.2.2 BiocNeighbors_2.0.1
#> [123] xml2_1.5.2 desc_1.4.3
#> [125] DelayedArray_0.32.0 plotly_4.11.0
#> [127] bookdown_0.44 checkmate_2.3.3
#> [129] scales_1.4.0 lmtest_0.9-40
#> [131] rappdirs_0.3.4 goftest_1.2-3
#> [133] stringr_1.6.0 digest_0.6.39
#> [135] spatstat.utils_3.2-1 rmarkdown_2.30
#> [137] basilisk_1.23.0 XVector_0.46.0
#> [139] htmltools_0.5.9 pkgconfig_2.0.3
#> [141] sparseMatrixStats_1.18.0 MatrixGenerics_1.18.1
#> [143] fastmap_1.2.0 rlang_1.1.7
#> [145] GlobalOptions_0.1.3 htmlwidgets_1.6.4
#> [147] UCSC.utils_1.2.0 shiny_1.12.1
#> [149] farver_2.1.2 jquerylib_0.1.4
#> [151] zoo_1.8-15 jsonlite_2.0.0
#> [153] BiocParallel_1.40.2 R.oo_1.27.1
#> [155] BiocSingular_1.22.0 magrittr_2.0.4
#> [157] scuttle_1.16.0 GenomeInfoDbData_1.2.13
#> [159] Rcpp_1.1.1 viridis_0.6.5
#> [161] reticulate_1.44.1 stringi_1.8.7
#> [163] zlibbioc_1.52.0 MASS_7.3-65
#> [165] plyr_1.8.9 parallel_4.4.0
#> [167] listenv_0.10.0 ggrepel_0.9.6
#> [169] deldir_2.0-4 splines_4.4.0
#> [171] tensor_1.5.1 hms_1.1.4
#> [173] circlize_0.4.17 locfit_1.5-9.12
#> [175] ps_1.9.1 spatstat.geom_3.6-1
#> [177] reshape2_1.4.5 stats4_4.4.0
#> [179] ScaledMatrix_1.14.0 XML_3.99-0.20
#> [181] evaluate_1.0.5 SeuratObject_4.1.4
#> [183] scran_1.34.0 BiocManager_1.30.27
#> [185] tzdb_0.5.0 foreach_1.5.2
#> [187] httpuv_1.6.16 polyclip_1.10-7
#> [189] RANN_2.6.2 purrr_1.2.1
#> [191] future_1.69.0 clue_0.3-66
#> [193] scattermore_1.2 rsvd_1.0.5
#> [195] xtable_1.8-4 later_1.4.5
#> [197] viridisLite_0.4.2 ragg_1.5.0
#> [199] tibble_3.3.1 websocket_1.4.4
#> [201] beeswarm_0.4.0 memoise_2.0.1
#> [203] IRanges_2.40.1 cluster_2.1.8.1
#> [205] timechange_0.3.0 globals_0.18.0