Introduction
CytoSPACER provides comprehensive visualization functions built on
ggplot2. This vignette showcases the various plotting
options available for exploring and presenting your results.
Generate Example Data
First, let’s create a simulated dataset:
set.seed(123)
# Parameters
n_genes <- 100
n_cells <- 500
n_spots <- 50
# Create scRNA-seq data
sc_data <- matrix(rpois(n_genes * n_cells, lambda = 5), nrow = n_genes, ncol = n_cells)
rownames(sc_data) <- paste0("Gene", seq_len(n_genes))
colnames(sc_data) <- paste0("Cell", seq_len(n_cells))
# Define cell types
cell_types <- rep(c("TypeA", "TypeB", "TypeC", "TypeD", "TypeE"), each = 100)
names(cell_types) <- colnames(sc_data)
# Add cell type-specific expression
for (i in 1:5) {
marker_genes <- ((i-1)*20 + 1):(i*20)
type_cells <- which(cell_types == unique(cell_types)[i])
sc_data[marker_genes, type_cells] <- sc_data[marker_genes, type_cells] + 20
}
# Create ST data
st_data <- matrix(rpois(n_genes * n_spots, lambda = 50), nrow = n_genes, ncol = n_spots)
rownames(st_data) <- paste0("Gene", seq_len(n_genes))
colnames(st_data) <- paste0("Spot", seq_len(n_spots))
# Create spatial coordinates (grid pattern)
coordinates <- data.frame(
row = rep(1:10, each = 5),
col = rep(1:5, times = 10),
row.names = colnames(st_data)
)
# Create cell type fractions
unique_types <- unique(cell_types)
n_types <- length(unique_types)
cell_type_fractions <- as.data.frame(matrix(
1/n_types, nrow = n_spots, ncol = n_types,
dimnames = list(colnames(st_data), unique_types)
))
# Run CytoSPACER
results <- run_cytospace(
sc_data = sc_data,
cell_types = cell_types,
st_data = st_data,
coordinates = coordinates,
cell_type_fractions = cell_type_fractions,
mean_cells_per_spot = 5,
seed = 42,
verbose = FALSE
)Basic Spatial Plots
Cell Type Distribution
The primary visualization shows the spatial distribution of assigned cell types:
plot_cytospace(results, type = "cell_types")
Customizing Point Size and Transparency
plot_cytospace(
results,
type = "cell_types",
point_size = 2.5,
alpha = 0.7
)
Adding Jitter
For dense regions where points overlap, add jitter:
plot_cytospace(
results,
type = "cell_types",
jitter = 0.3,
point_size = 2
)
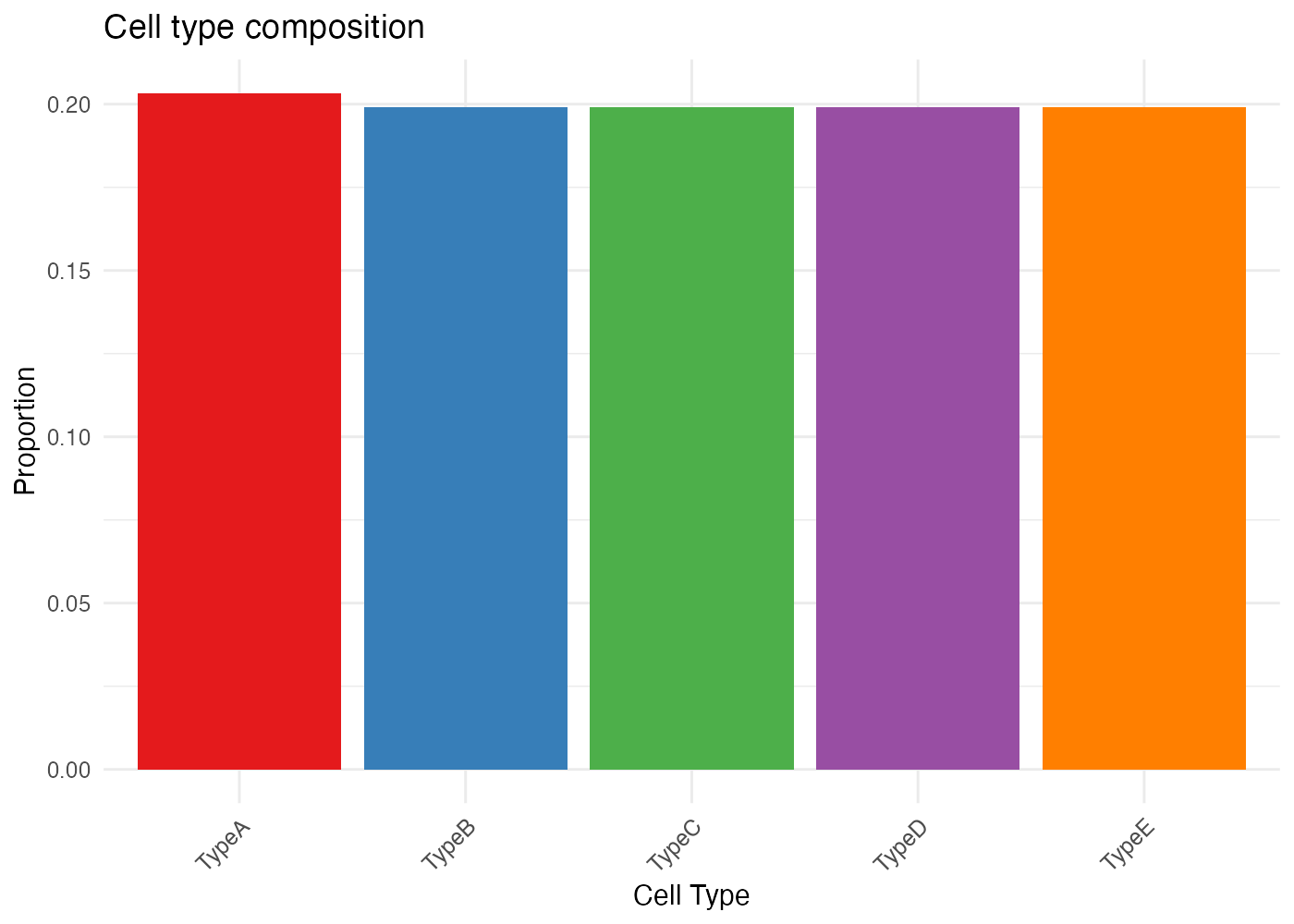
Custom Colors
my_colors <- c(
"TypeA" = "#E41A1C",
"TypeB" = "#377EB8",
"TypeC" = "#4DAF4A",
"TypeD" = "#984EA3",
"TypeE" = "#FF7F00"
)
plot_cytospace(
results,
type = "cell_types",
colors = my_colors,
point_size = 2.5
)
Custom Title
plot_cytospace(
results,
type = "cell_types",
title = "Spatial Distribution of Cell Types",
point_size = 2
)
Advanced Visualizations
Combining with ggplot2
Since all plots are ggplot2 objects, you can easily customize them:
p <- plot_cytospace(results, type = "cell_types", colors = my_colors)
# Add custom theme
p +
theme_minimal() +
theme(
legend.position = "bottom",
legend.title = element_text(face = "bold"),
plot.title = element_text(hjust = 0.5, size = 16, face = "bold")
) +
guides(color = guide_legend(nrow = 1, override.aes = list(size = 4)))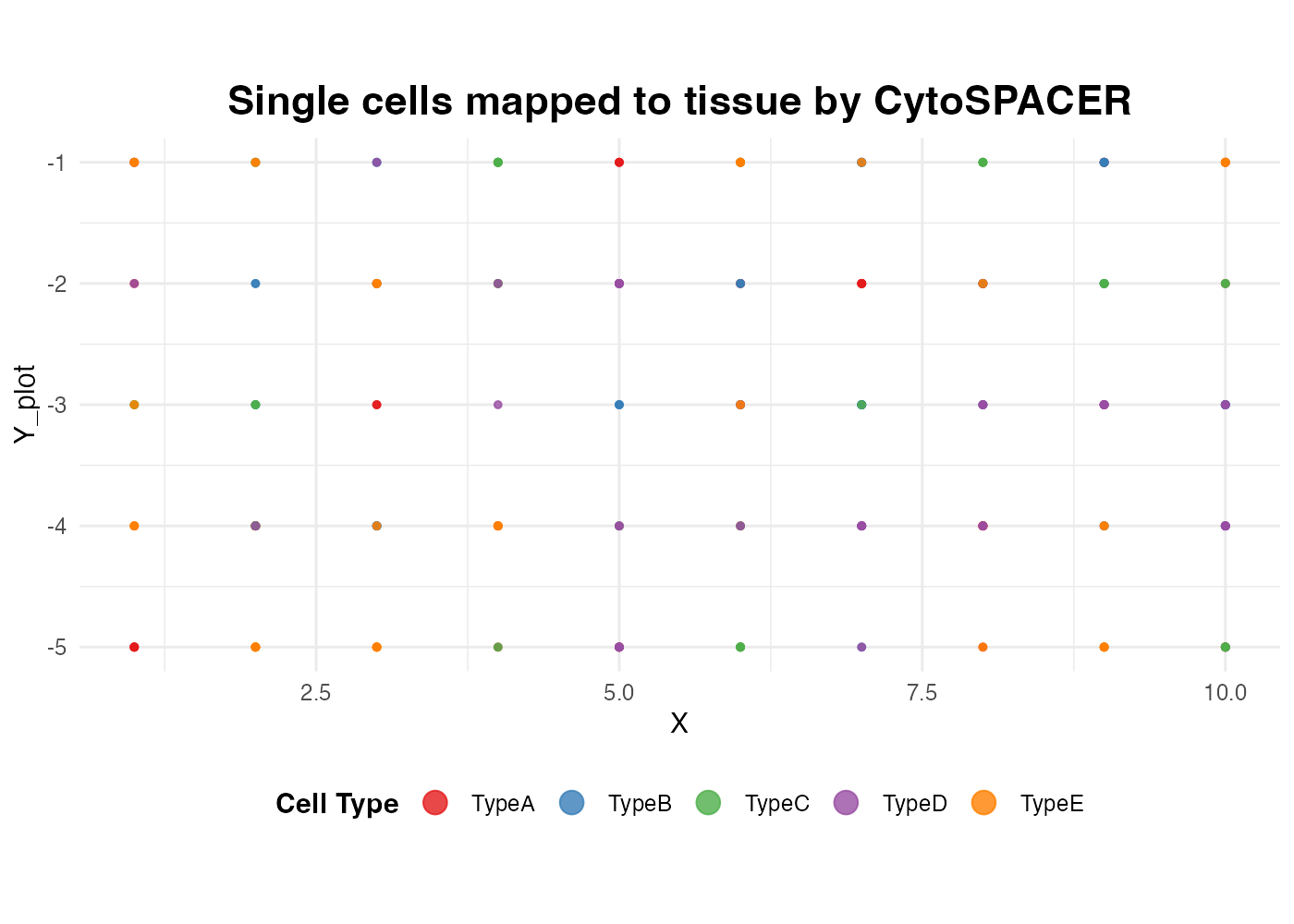
Faceted by Cell Type
# Get the assigned locations data
locs <- results$assigned_locations
locs$Y_plot <- -locs$col
# Create faceted plot
ggplot(locs, aes(x = row, y = Y_plot)) +
geom_point(aes(color = CellType), size = 1.5, alpha = 0.8) +
facet_wrap(~ CellType, ncol = 3) +
scale_color_manual(values = my_colors) +
coord_equal() +
theme_minimal() +
theme(
axis.text = element_blank(),
axis.title = element_blank(),
axis.ticks = element_blank(),
panel.grid = element_blank(),
strip.text = element_text(face = "bold", size = 11),
legend.position = "none"
) +
labs(title = "Cell Type Distribution by Type")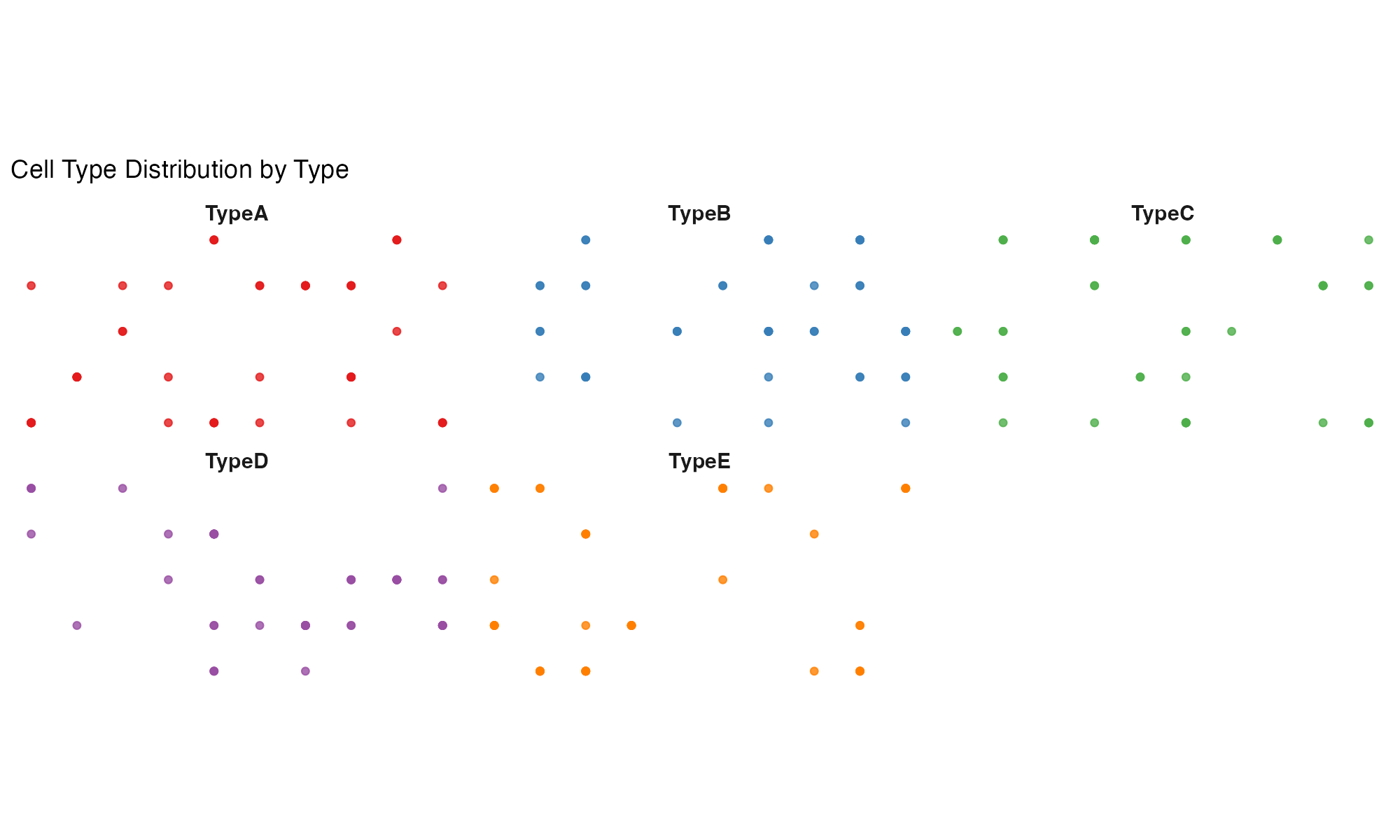
Saving Plots
# Save individual plot
p <- plot_cytospace(results, type = "cell_types", colors = my_colors)
# Using CytoSPACER function
save_cytospace_plot(
plot = p,
output_dir = "figures/",
prefix = "spatial_",
formats = c("png", "pdf"),
width = 10,
height = 8,
dpi = 300
)
# Using ggplot2 directly
ggsave("figures/cell_types.png", p, width = 10, height = 8, dpi = 300)Color Palettes
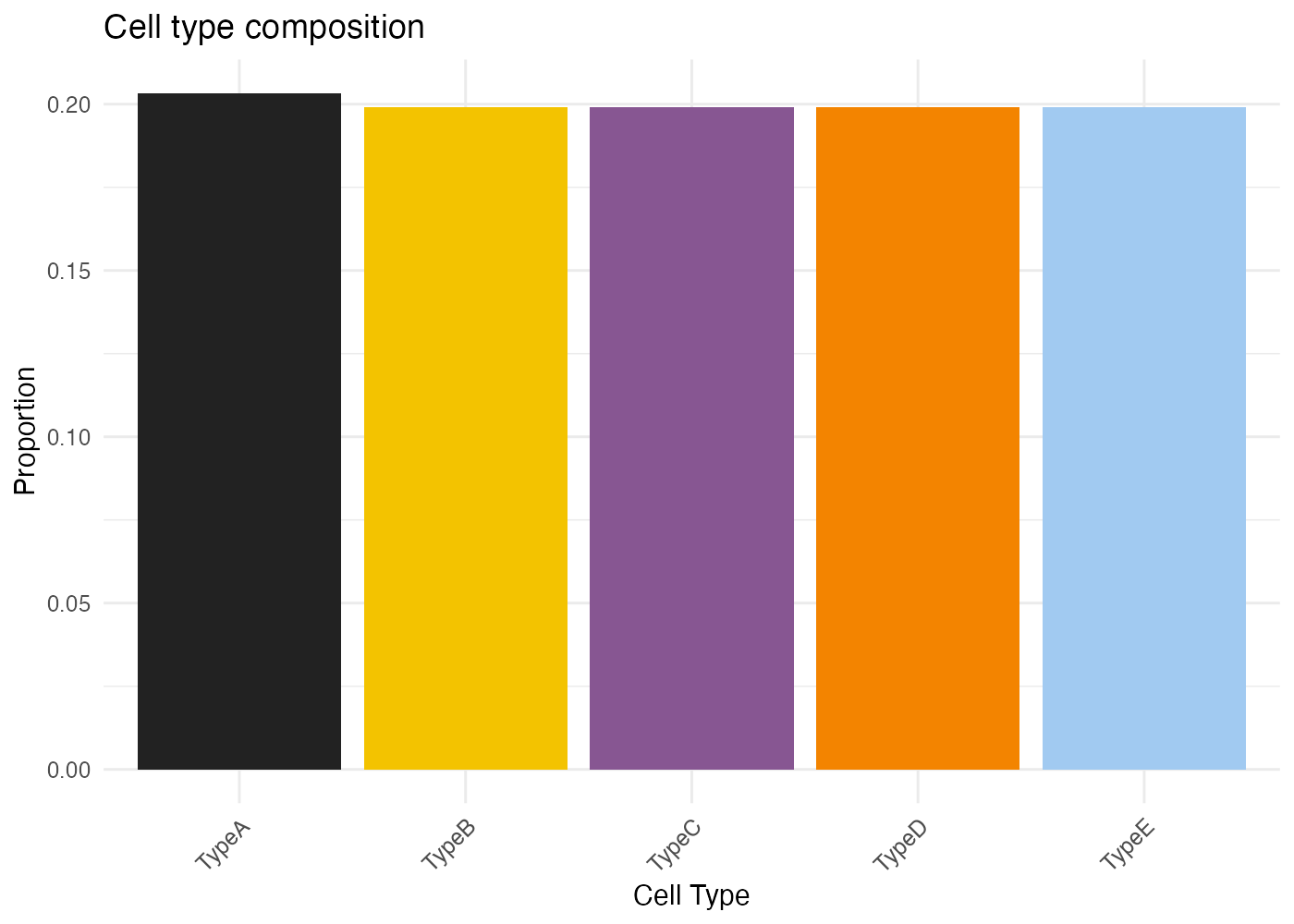
CytoSPACER includes a built-in color palette optimized for distinguishing cell types:
# Default CytoSPACER colors (first 10)
default_colors <- c(
"#222222", "#F3C300", "#875692", "#F38400", "#A1CAF1",
"#BE0032", "#C2B280", "#848482", "#008856", "#E68FAC"
)
# Display palette
barplot(rep(1, 10), col = default_colors, border = NA, axes = FALSE,
main = "CytoSPACER Default Color Palette", names.arg = 1:10)
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggplot2_4.0.1 CytoSPACER_1.0.0
#>
#> loaded via a namespace (and not attached):
#> [1] sass_0.4.10 future_1.69.0 generics_0.1.4
#> [4] lattice_0.22-7 listenv_0.10.0 digest_0.6.39
#> [7] magrittr_2.0.4 evaluate_1.0.5 grid_4.4.0
#> [10] RColorBrewer_1.1-3 fastmap_1.2.0 jsonlite_2.0.0
#> [13] Matrix_1.7-4 scales_1.4.0 codetools_0.2-20
#> [16] textshaping_1.0.4 jquerylib_0.1.4 cli_3.6.5
#> [19] rlang_1.1.7 parallelly_1.46.1 future.apply_1.20.1
#> [22] withr_3.0.2 cachem_1.1.0 yaml_2.3.12
#> [25] otel_0.2.0 tools_4.4.0 parallel_4.4.0
#> [28] dplyr_1.1.4 globals_0.18.0 vctrs_0.7.1
#> [31] R6_2.6.1 lifecycle_1.0.5 fs_1.6.6
#> [34] htmlwidgets_1.6.4 ragg_1.5.0 pkgconfig_2.0.3
#> [37] desc_1.4.3 pkgdown_2.1.3 progressr_0.18.0
#> [40] bslib_0.9.0 pillar_1.11.1 gtable_0.3.6
#> [43] data.table_1.18.0 glue_1.8.0 Rcpp_1.1.1
#> [46] systemfonts_1.3.1 xfun_0.56 tibble_3.3.1
#> [49] tidyselect_1.2.1 knitr_1.51 dichromat_2.0-0.1
#> [52] farver_2.1.2 htmltools_0.5.9 rmarkdown_2.30
#> [55] labeling_0.4.3 compiler_4.4.0 S7_0.2.1