Bulk RNA-seq Metabolic Flux Analysis
Zaoqu Liu
2026-01-23
Source:vignettes/bulk_analysis.Rmd
bulk_analysis.RmdOverview
This vignette demonstrates the complete workflow for metabolic flux analysis from bulk RNA-seq data using METAFLUX.
Load Package and Data
library(METAFLUX)
library(ggplot2)
# Load example bulk RNA-seq data
data("bulk_test_example")
data("human_blood")
data("human_gem")
# Inspect data
cat("Gene expression matrix:\n")
#> Gene expression matrix:
cat(" Dimensions:", dim(bulk_test_example), "\n")
#> Dimensions: 58581 5
cat(" Samples:", colnames(bulk_test_example), "\n")
#> Samples: Sample1 Sample2 Sample3 Sample4 Sample5
cat(" Example genes:", head(rownames(bulk_test_example), 5), "\n")
#> Example genes: TSPAN6 TNMD DPM1 SCYL3 C1orf112Step 1: Calculate MRAS
Metabolic Reaction Activity Scores (MRAS) integrate gene expression with metabolic network topology.
# Calculate MRAS
mras <- calculate_reaction_score(bulk_test_example)
# Check output
cat("\nMRAS matrix:\n")
#>
#> MRAS matrix:
cat(" Dimensions:", dim(mras), "\n")
#> Dimensions: 13082 5
cat(" Score range:", range(as.matrix(mras)), "\n")
#> Score range: 0 1MRAS Distribution
mras_values <- as.vector(as.matrix(mras))
mras_df <- data.frame(MRAS = mras_values[mras_values > 0])
ggplot(mras_df, aes(x = MRAS)) +
geom_histogram(bins = 50, fill = "#3498db", color = "white", alpha = 0.8) +
labs(
title = "Distribution of Metabolic Reaction Activity Scores",
x = "MRAS Value",
y = "Count"
) +
theme_minimal() +
theme(plot.title = element_text(hjust = 0.5, face = "bold"))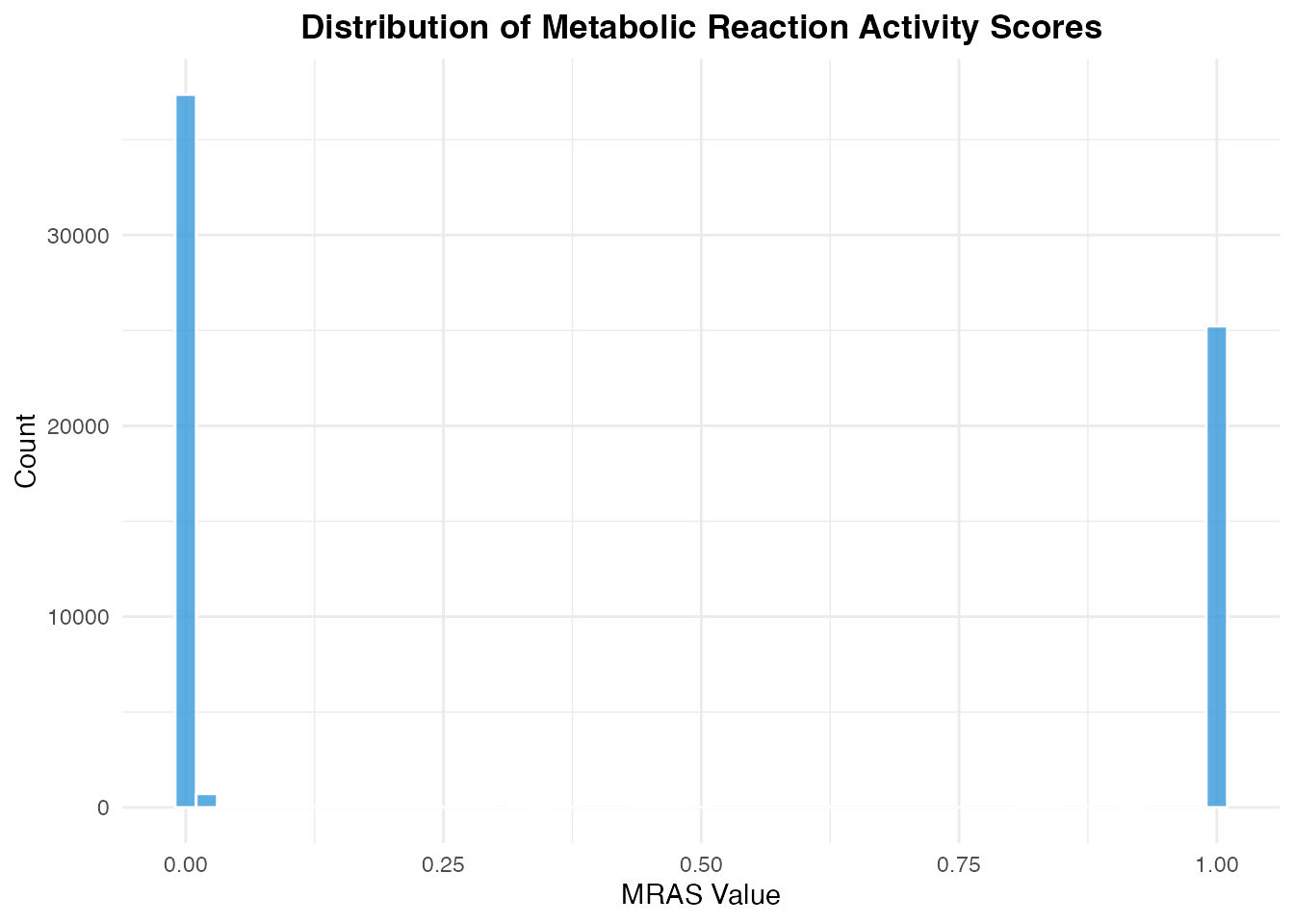
Distribution of MRAS values
Step 2: Compute Metabolic Fluxes
# Run flux balance analysis
flux <- compute_flux(mras = mras, medium = human_blood)
#> | | | 0% | |============== | 20% | |============================ | 40% | |========================================== | 60% | |======================================================== | 80% | |======================================================================| 100%
# Check results
cat("\nFlux matrix:\n")
#>
#> Flux matrix:
cat(" Dimensions:", dim(flux), "\n")
#> Dimensions: 13082 5
cat(" Flux range:", range(flux), "\n")
#> Flux range: -0.08404168 0.08406293Flux Interpretation
# Classify reactions by flux direction
flux_mean <- rowMeans(flux)
flux_class <- data.frame(
Direction = c("Production (+)", "Consumption (-)", "Inactive (≈0)"),
Count = c(
sum(flux_mean > 0.001),
sum(flux_mean < -0.001),
sum(abs(flux_mean) <= 0.001)
)
)
ggplot(flux_class, aes(x = Direction, y = Count, fill = Direction)) +
geom_bar(stat = "identity", width = 0.6) +
geom_text(aes(label = Count), vjust = -0.5, size = 5) +
scale_fill_manual(values = c("#27ae60", "#e74c3c", "#95a5a6")) +
labs(
title = "Reaction Classification by Flux Direction",
x = "",
y = "Number of Reactions"
) +
theme_minimal() +
theme(
plot.title = element_text(hjust = 0.5, face = "bold"),
legend.position = "none"
) +
ylim(0, max(flux_class$Count) * 1.1)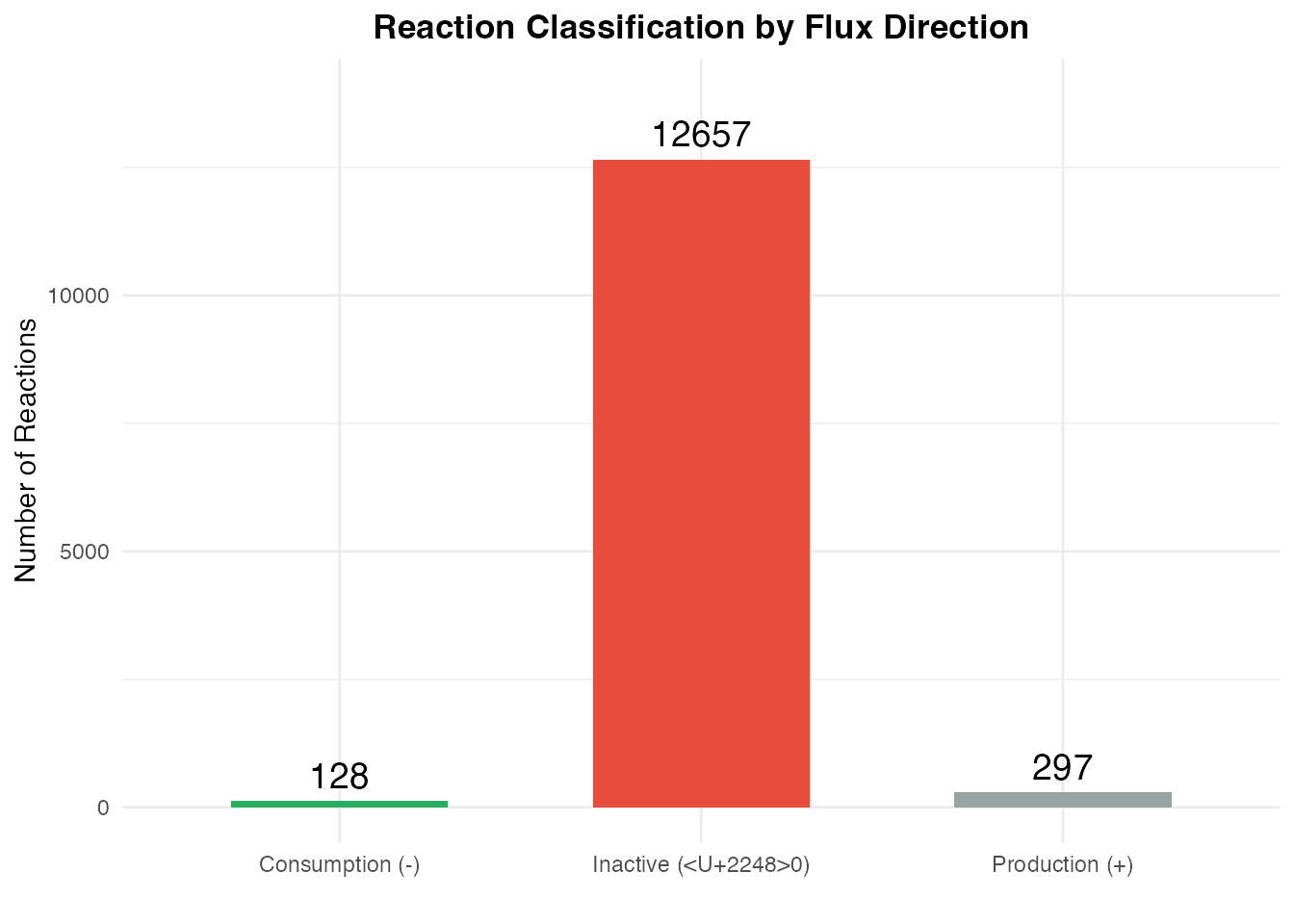
Interpretation of flux values
Step 3: Key Metabolic Indicators
Central Carbon Metabolism
# Define key reactions
central_carbon <- c(
"Glucose uptake" = "HMR_9034",
"Lactate secretion" = "HMR_9135",
"Pyruvate transport" = "HMR_9133",
"Glutamine uptake" = "HMR_9063"
)
# Extract and plot
cc_flux <- flux[central_carbon, , drop = FALSE]
rownames(cc_flux) <- names(central_carbon)
cc_df <- data.frame(
Reaction = rep(rownames(cc_flux), ncol(cc_flux)),
Sample = rep(colnames(cc_flux), each = nrow(cc_flux)),
Flux = as.vector(as.matrix(cc_flux))
)
ggplot(cc_df, aes(x = Reaction, y = Flux, fill = Sample)) +
geom_bar(stat = "identity", position = "dodge") +
geom_hline(yintercept = 0, linetype = "dashed") +
scale_fill_brewer(palette = "Set2") +
labs(
title = "Central Carbon Metabolism",
subtitle = "Key reaction fluxes across samples",
x = "",
y = "Flux"
) +
theme_minimal() +
theme(
plot.title = element_text(hjust = 0.5, face = "bold"),
axis.text.x = element_text(angle = 30, hjust = 1)
)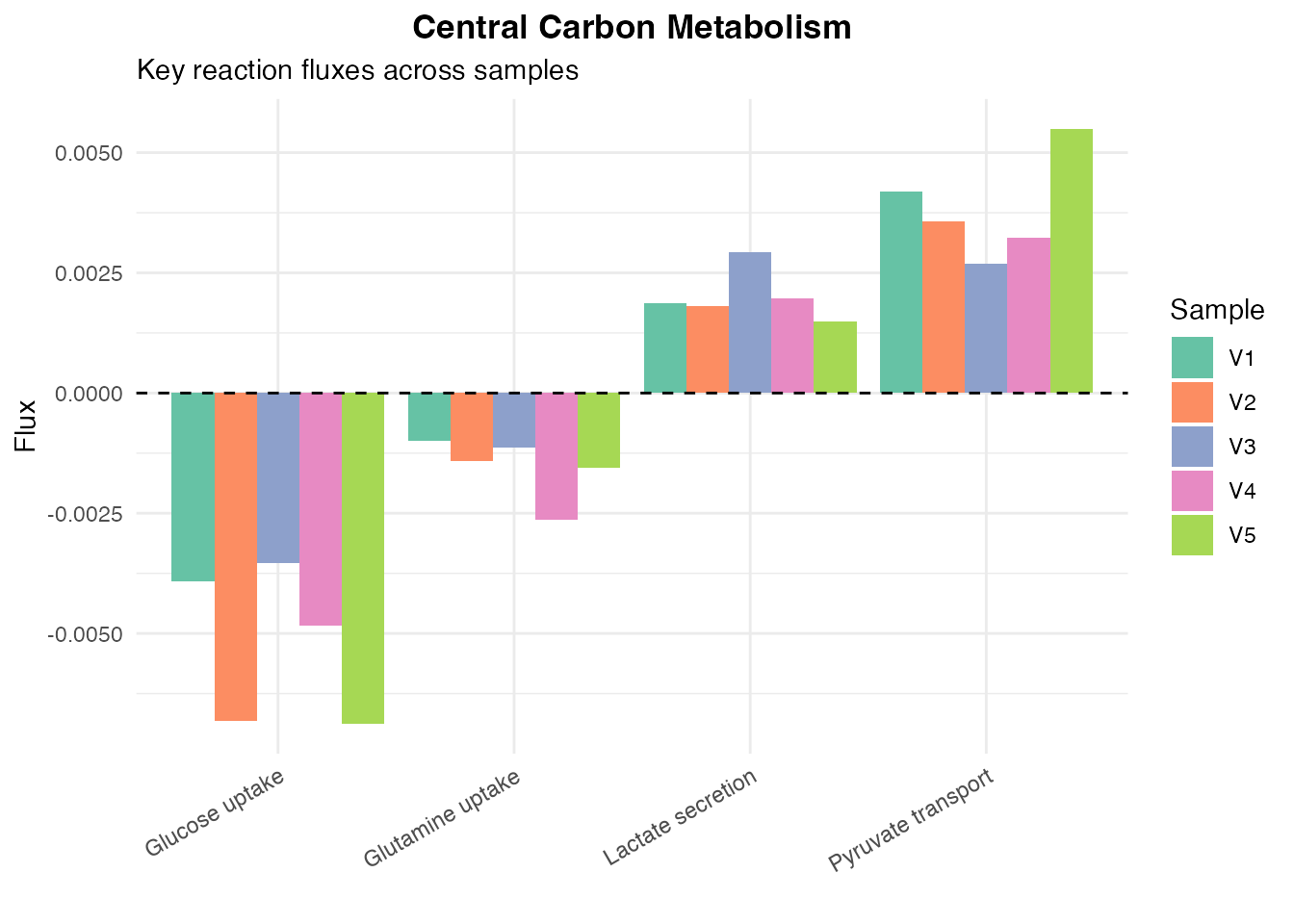
Central carbon metabolism flux
Biomass Production
biomass_flux <- flux["biomass_human", ]
biomass_df <- data.frame(
Sample = names(biomass_flux),
Flux = as.numeric(biomass_flux)
)
ggplot(biomass_df, aes(x = Sample, y = Flux, fill = Sample)) +
geom_bar(stat = "identity", width = 0.6) +
scale_fill_brewer(palette = "Set2") +
labs(
title = "Biomass Production Rate",
subtitle = "Proxy for cellular growth rate",
x = "Sample",
y = "Biomass Flux"
) +
theme_minimal() +
theme(
plot.title = element_text(hjust = 0.5, face = "bold"),
legend.position = "none"
)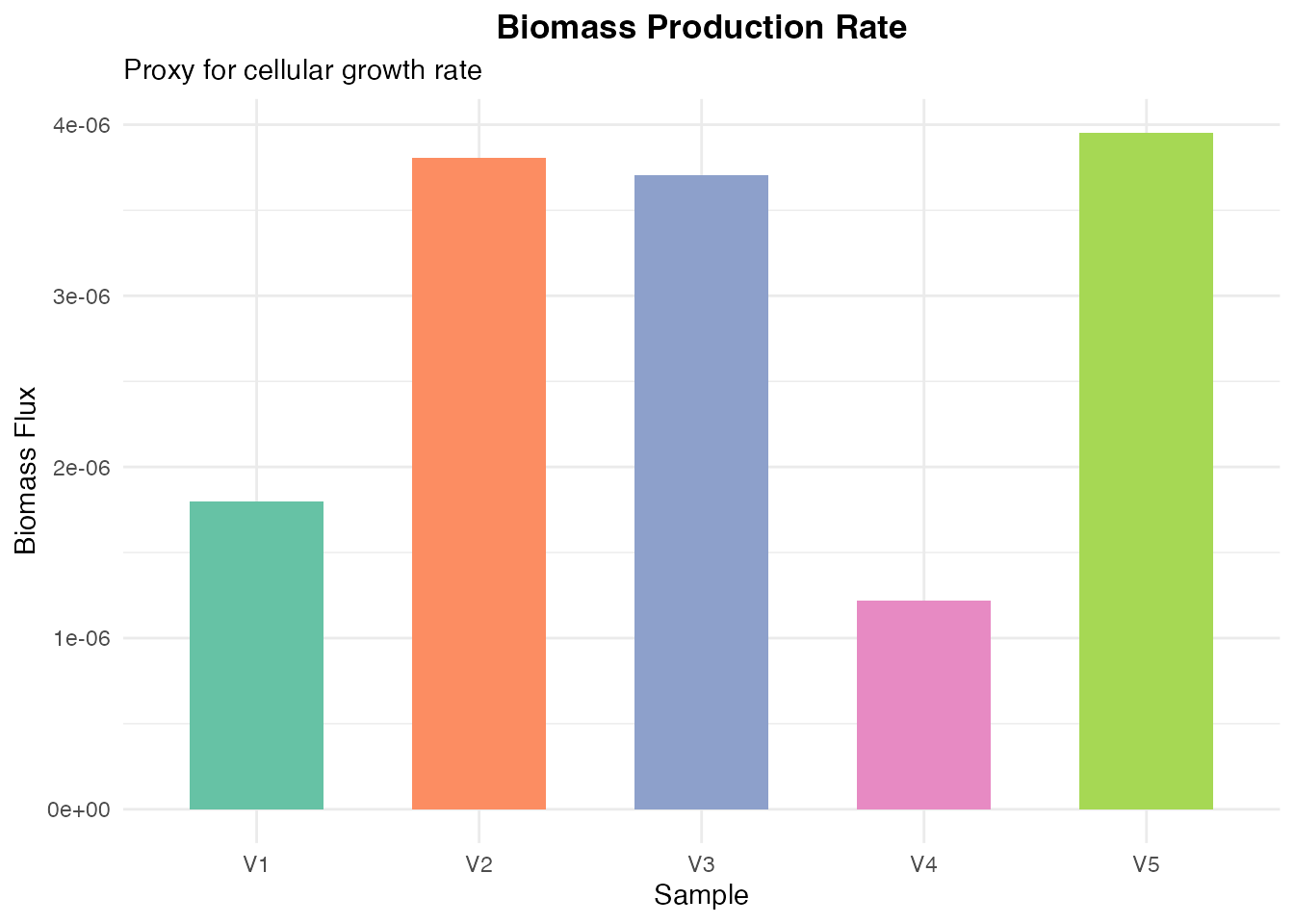
Biomass production flux (growth rate proxy)
Step 4: Pathway Analysis
# Map reactions to pathways
pathway_map <- human_gem$SUBSYSTEM
names(pathway_map) <- human_gem$ID
# Calculate pathway activity
pathways <- unique(pathway_map)
pathway_activity <- sapply(pathways, function(pw) {
rxns <- names(pathway_map)[pathway_map == pw]
rxns <- intersect(rxns, rownames(flux))
if (length(rxns) > 0) mean(abs(flux[rxns, ])) else NA
})
# Top 12 pathways
top_pw <- sort(pathway_activity[!is.na(pathway_activity)], decreasing = TRUE)[1:12]
pw_df <- data.frame(
Pathway = factor(names(top_pw), levels = rev(names(top_pw))),
Activity = top_pw
)
ggplot(pw_df, aes(x = Activity, y = Pathway)) +
geom_bar(stat = "identity", fill = "#2c3e50") +
labs(
title = "Top 12 Active Metabolic Pathways",
x = "Mean Absolute Flux",
y = ""
) +
theme_minimal() +
theme(
plot.title = element_text(hjust = 0.5, face = "bold"),
axis.text.y = element_text(size = 9)
)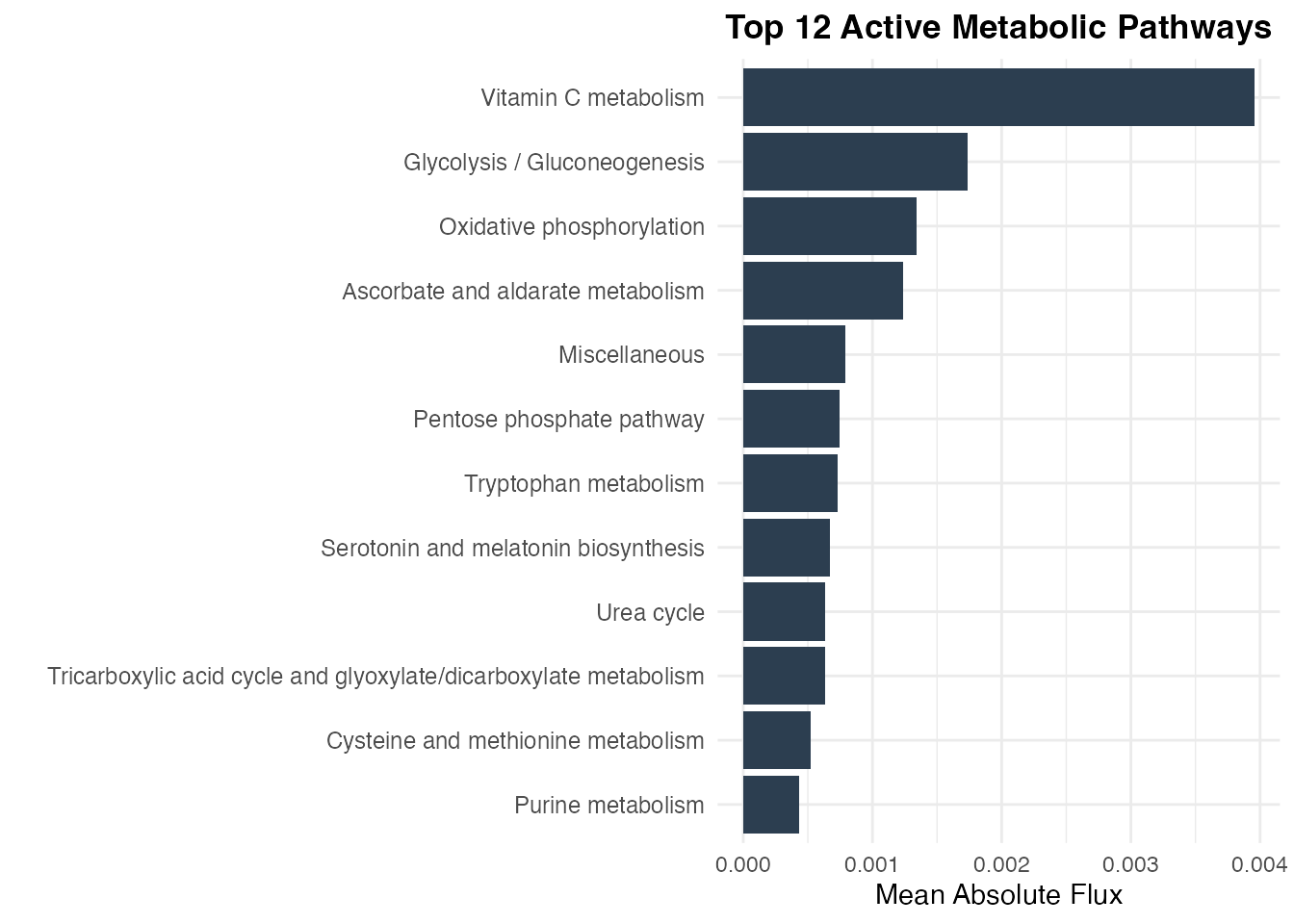
Top metabolic pathways by activity
Step 5: Quality Control
Steady-State Verification
# Load stoichiometric matrix
Hgem <- METAFLUX:::Hgem
# Check steady-state constraint: S*v = 0
sv_violations <- sapply(1:ncol(flux), function(i) {
sv <- Hgem$S %*% flux[, i]
max(abs(sv))
})
cat("Steady-state constraint check:\n")
#> Steady-state constraint check:
cat(" Max violations per sample:", round(sv_violations, 6), "\n")
#> Max violations per sample: 2.3e-05 7e-06 1.4e-05 1.7e-05 1.3e-05
cat(" All < 0.001:", all(sv_violations < 0.001), "\n")
#> All < 0.001: TRUEGene Coverage
gene_num <- METAFLUX:::gene_num
input_genes <- rownames(bulk_test_example)
metabolic_genes <- rownames(gene_num)
coverage <- sum(input_genes %in% metabolic_genes)
total <- length(metabolic_genes)
cov_df <- data.frame(
Category = c("Detected", "Missing"),
Count = c(coverage, total - coverage)
)
ggplot(cov_df, aes(x = "", y = Count, fill = Category)) +
geom_bar(stat = "identity", width = 1) +
coord_polar("y") +
scale_fill_manual(values = c("Detected" = "#27ae60", "Missing" = "#e74c3c")) +
labs(
title = "Metabolic Gene Coverage",
subtitle = sprintf("%.1f%% (%d/%d genes)", coverage/total*100, coverage, total)
) +
theme_void() +
theme(
plot.title = element_text(hjust = 0.5, face = "bold"),
plot.subtitle = element_text(hjust = 0.5)
)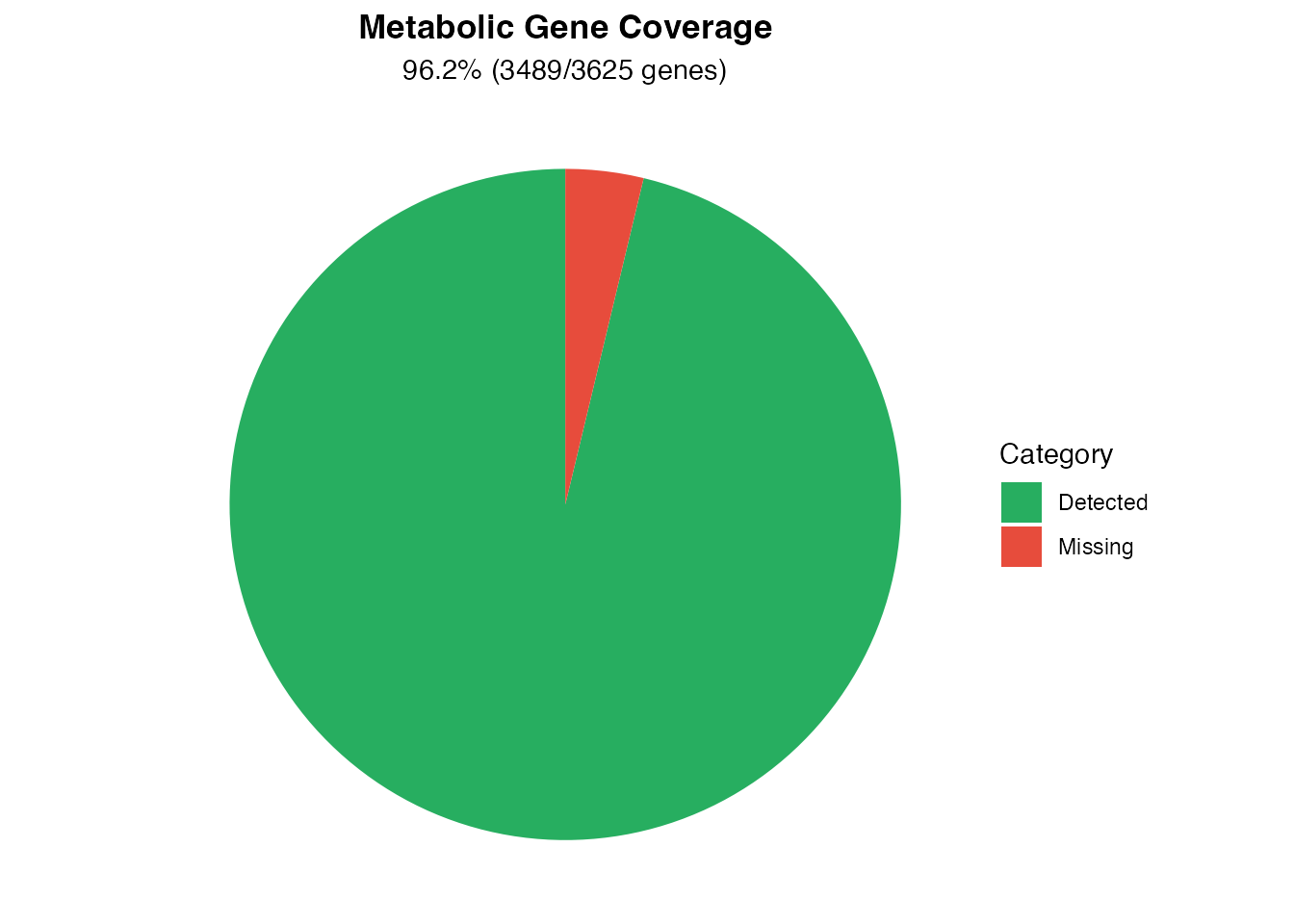
Metabolic gene coverage
Summary
cat("================================================\n")
#> ================================================
cat("METAFLUX Bulk RNA-seq Analysis Complete\n")
#> METAFLUX Bulk RNA-seq Analysis Complete
cat("================================================\n\n")
#> ================================================
cat("Input:\n")
#> Input:
cat(sprintf(" Genes: %d\n", nrow(bulk_test_example)))
#> Genes: 58581
cat(sprintf(" Samples: %d\n", ncol(bulk_test_example)))
#> Samples: 5
cat(sprintf(" Metabolic gene coverage: %.1f%%\n", coverage/total*100))
#> Metabolic gene coverage: 96.2%
cat("\nOutput:\n")
#>
#> Output:
cat(sprintf(" Reactions: %d\n", nrow(flux)))
#> Reactions: 13082
cat(sprintf(" Flux range: [%.4f, %.4f]\n", min(flux), max(flux)))
#> Flux range: [-0.0840, 0.0841]
cat("\nKey findings:\n")
#>
#> Key findings:
cat(sprintf(" Active reactions: %d\n", sum(abs(rowMeans(flux)) > 0.001)))
#> Active reactions: 425
cat(sprintf(" Glucose uptake (mean): %.4f\n", mean(flux["HMR_9034", ])))
#> Glucose uptake (mean): -0.0052
cat(sprintf(" Lactate secretion (mean): %.4f\n", mean(flux["HMR_9135", ])))
#> Lactate secretion (mean): 0.0020Author
Zaoqu Liu - Package maintainer and developer
- GitHub: Zaoqu-Liu
- ORCID: 0000-0002-0452-742X
- Email: liuzaoqu@163.com
Session Information
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggplot2_4.0.1 METAFLUX_2.2.0
#>
#> loaded via a namespace (and not attached):
#> [1] deldir_2.0-4 pbapply_1.7-4 gridExtra_2.3
#> [4] rlang_1.1.7 magrittr_2.0.4 RcppAnnoy_0.0.23
#> [7] otel_0.2.0 spatstat.geom_3.6-1 matrixStats_1.5.0
#> [10] ggridges_0.5.7 compiler_4.4.0 png_0.1-8
#> [13] systemfonts_1.3.1 vctrs_0.7.0 reshape2_1.4.5
#> [16] stringr_1.6.0 pkgconfig_2.0.3 fastmap_1.2.0
#> [19] labeling_0.4.3 promises_1.5.0 rmarkdown_2.30
#> [22] ragg_1.5.0 purrr_1.2.1 xfun_0.56
#> [25] cachem_1.1.0 jsonlite_2.0.0 goftest_1.2-3
#> [28] later_1.4.5 spatstat.utils_3.2-1 irlba_2.3.5.1
#> [31] parallel_4.4.0 cluster_2.1.8.1 R6_2.6.1
#> [34] ica_1.0-3 spatstat.data_3.1-9 bslib_0.9.0
#> [37] stringi_1.8.7 RColorBrewer_1.1-3 reticulate_1.44.1
#> [40] spatstat.univar_3.1-6 parallelly_1.46.1 lmtest_0.9-40
#> [43] jquerylib_0.1.4 scattermore_1.2 iterators_1.0.14
#> [46] Rcpp_1.1.1 knitr_1.51 tensor_1.5.1
#> [49] future.apply_1.20.1 zoo_1.8-15 sctransform_0.4.3
#> [52] httpuv_1.6.16 Matrix_1.7-4 splines_4.4.0
#> [55] igraph_2.2.1 tidyselect_1.2.1 abind_1.4-8
#> [58] dichromat_2.0-0.1 yaml_2.3.12 doParallel_1.0.17
#> [61] spatstat.random_3.4-3 spatstat.explore_3.6-0 codetools_0.2-20
#> [64] miniUI_0.1.2 listenv_0.10.0 plyr_1.8.9
#> [67] lattice_0.22-7 tibble_3.3.1 withr_3.0.2
#> [70] shiny_1.12.1 S7_0.2.1 ROCR_1.0-11
#> [73] evaluate_1.0.5 Rtsne_0.17 future_1.69.0
#> [76] desc_1.4.3 survival_3.8-3 polyclip_1.10-7
#> [79] fitdistrplus_1.2-4 osqp_0.6.3.3 pillar_1.11.1
#> [82] Seurat_4.4.0 KernSmooth_2.23-26 foreach_1.5.2
#> [85] plotly_4.11.0 generics_0.1.4 sp_2.2-0
#> [88] scales_1.4.0 globals_0.18.0 xtable_1.8-4
#> [91] glue_1.8.0 lazyeval_0.2.2 tools_4.4.0
#> [94] data.table_1.18.0 RANN_2.6.2 fs_1.6.6
#> [97] leiden_0.4.3.1 cowplot_1.2.0 grid_4.4.0
#> [100] tidyr_1.3.2 nlme_3.1-168 patchwork_1.3.2
#> [103] cli_3.6.5 spatstat.sparse_3.1-0 textshaping_1.0.4
#> [106] viridisLite_0.4.2 dplyr_1.1.4 uwot_0.2.4
#> [109] gtable_0.3.6 sass_0.4.10 digest_0.6.39
#> [112] progressr_0.18.0 ggrepel_0.9.6 htmlwidgets_1.6.4
#> [115] SeuratObject_4.1.4 farver_2.1.2 htmltools_0.5.9
#> [118] pkgdown_2.1.3 lifecycle_1.0.5 httr_1.4.7
#> [121] mime_0.13 MASS_7.3-65