Visualization Guide for METAFLUX
Zaoqu Liu
2026-01-23
Source:vignettes/visualization.Rmd
visualization.RmdStep 1: Calculate MRAS
# Calculate Metabolic Reaction Activity Scores
mras <- calculate_reaction_score(bulk_test_example)
cat("MRAS matrix:", nrow(mras), "reactions x", ncol(mras), "samples\n")
#> MRAS matrix: 13082 reactions x 5 samplesStep 2: Compute Fluxes
# Compute metabolic fluxes
flux <- compute_flux(mras = mras, medium = human_blood)
#> | | | 0% | |============== | 20% | |============================ | 40% | |========================================== | 60% | |======================================================== | 80% | |======================================================================| 100%
cat("Flux matrix:", nrow(flux), "reactions x", ncol(flux), "samples\n")
#> Flux matrix: 13082 reactions x 5 samplesVisualization 1: Flux Distribution
# Prepare data
flux_values <- as.vector(as.matrix(flux))
flux_df <- data.frame(flux = flux_values[abs(flux_values) < 0.5])
# Plot
ggplot(flux_df, aes(x = flux)) +
geom_histogram(bins = 100, fill = "#3498db", color = "white", alpha = 0.8) +
labs(
title = "Distribution of Metabolic Fluxes",
subtitle = "METAFLUX analysis of bulk RNA-seq data",
x = "Flux Value",
y = "Count"
) +
theme_minimal() +
theme(
plot.title = element_text(hjust = 0.5, size = 14, face = "bold"),
plot.subtitle = element_text(hjust = 0.5, size = 10, color = "gray50")
)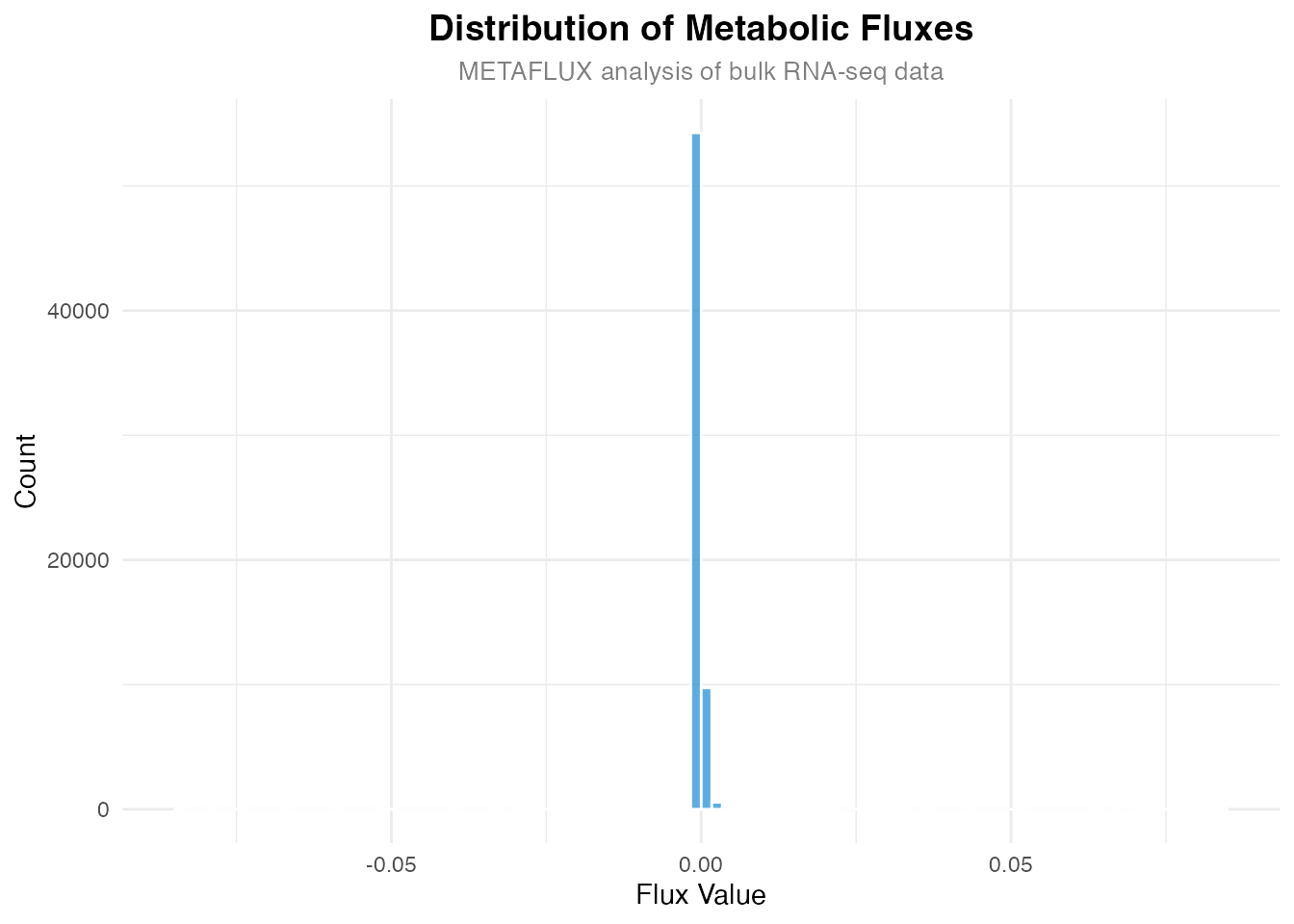
Distribution of metabolic fluxes across all samples
Visualization 2: Key Metabolite Exchange
# Define key reactions
key_rxns <- c(
"Glucose" = "HMR_9034",
"Lactate" = "HMR_9135",
"Glutamine" = "HMR_9063",
"Pyruvate" = "HMR_9133"
)
# Extract flux values
key_flux <- flux[key_rxns, , drop = FALSE]
rownames(key_flux) <- names(key_rxns)
# Convert to long format
key_df <- data.frame(
Metabolite = rep(rownames(key_flux), ncol(key_flux)),
Sample = rep(colnames(key_flux), each = nrow(key_flux)),
Flux = as.vector(as.matrix(key_flux))
)
# Plot
ggplot(key_df, aes(x = Metabolite, y = Flux, fill = Sample)) +
geom_bar(stat = "identity", position = "dodge", width = 0.7) +
geom_hline(yintercept = 0, linetype = "dashed", color = "gray50") +
labs(
title = "Key Metabolite Exchange",
subtitle = "Negative = uptake, Positive = secretion",
x = "",
y = "Flux"
) +
scale_fill_brewer(palette = "Set2") +
theme_minimal() +
theme(
plot.title = element_text(hjust = 0.5, size = 14, face = "bold"),
plot.subtitle = element_text(hjust = 0.5, size = 10, color = "gray50"),
axis.text.x = element_text(angle = 45, hjust = 1, size = 11)
)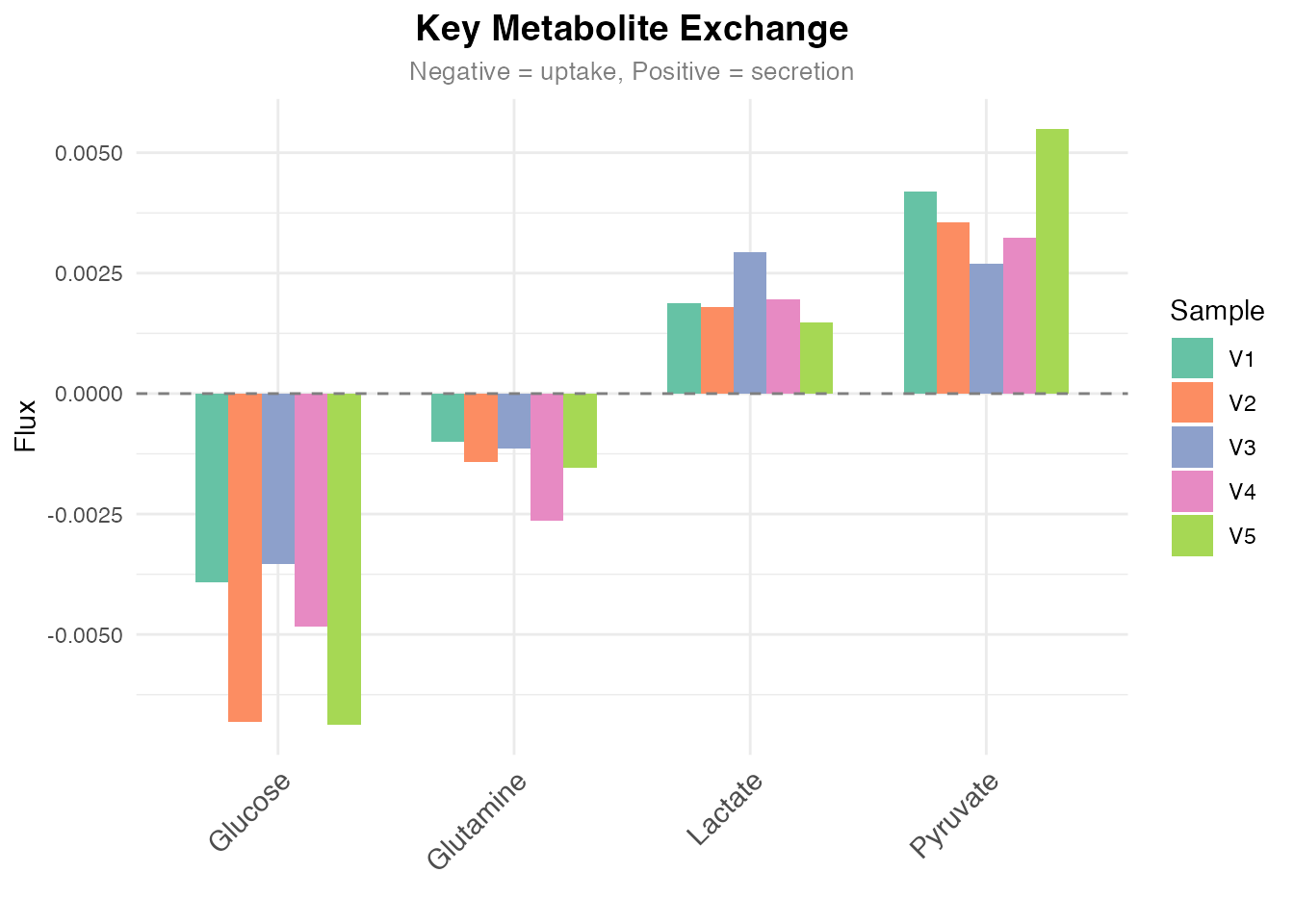
Flux values for key metabolic reactions
Visualization 3: Top Variable Reactions
# Calculate variance
flux_var <- apply(flux, 1, var)
top_30 <- names(sort(flux_var, decreasing = TRUE)[1:30])
# Prepare matrix
flux_top <- as.matrix(flux[top_30, ])
# Scale for visualization
flux_scaled <- t(scale(t(flux_top)))
# Convert to data frame
heatmap_df <- data.frame(
Reaction = rep(rownames(flux_scaled), ncol(flux_scaled)),
Sample = rep(colnames(flux_scaled), each = nrow(flux_scaled)),
Flux = as.vector(flux_scaled)
)
# Order reactions by mean flux
rxn_order <- rownames(flux_scaled)[order(rowMeans(flux_scaled))]
heatmap_df$Reaction <- factor(heatmap_df$Reaction, levels = rxn_order)
# Plot heatmap
ggplot(heatmap_df, aes(x = Sample, y = Reaction, fill = Flux)) +
geom_tile() +
scale_fill_gradient2(
low = "#2166ac", mid = "white", high = "#b2182b",
midpoint = 0, name = "Z-score"
) +
labs(
title = "Top 30 Variable Metabolic Reactions",
x = "Sample",
y = "Reaction"
) +
theme_minimal() +
theme(
plot.title = element_text(hjust = 0.5, size = 14, face = "bold"),
axis.text.y = element_text(size = 6),
axis.text.x = element_text(angle = 45, hjust = 1)
)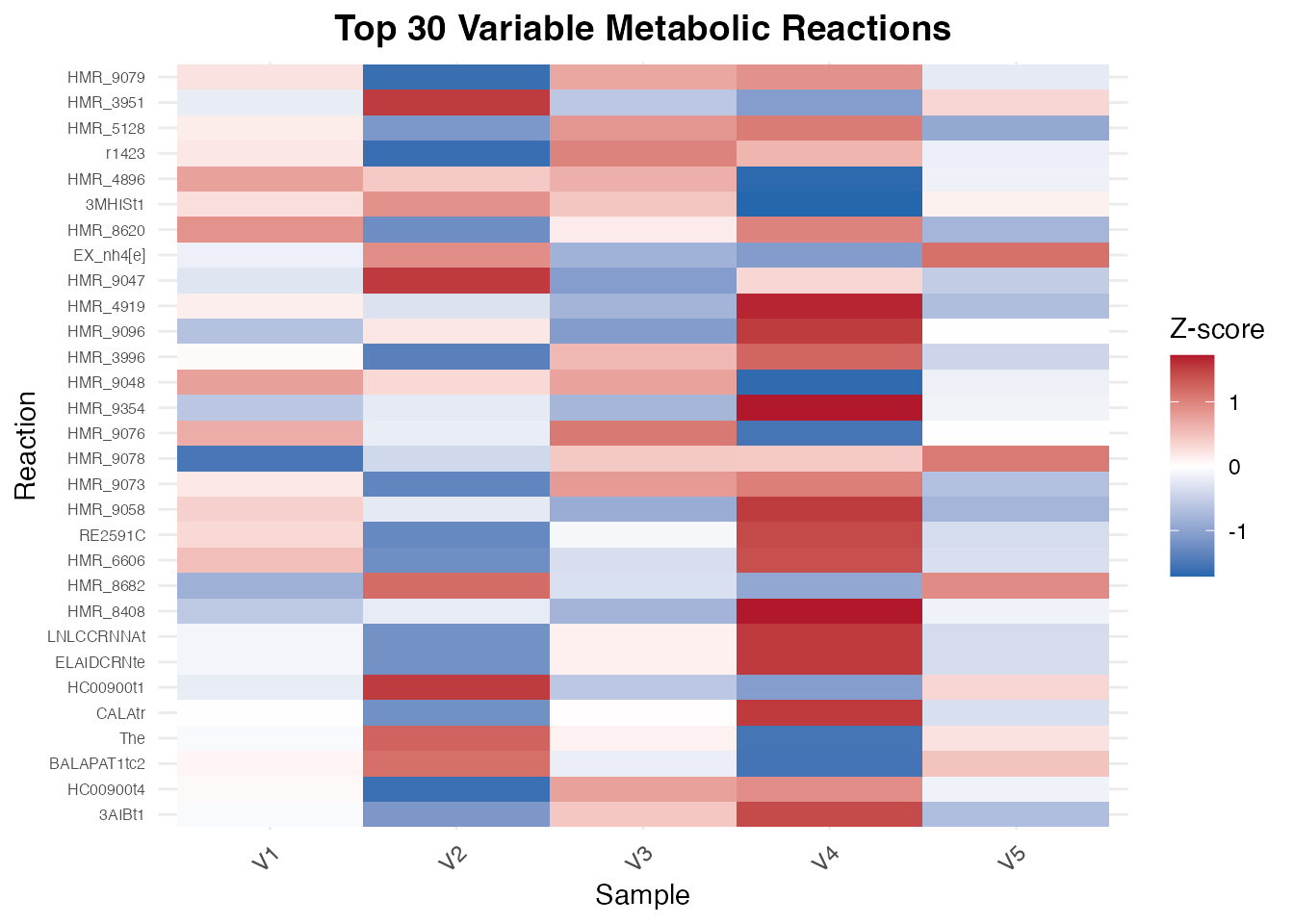
Heatmap of top 30 most variable metabolic reactions
Visualization 4: Pathway Activity
# Get pathway information
pathway_map <- human_gem$SUBSYSTEM
names(pathway_map) <- human_gem$ID
# Calculate mean flux per pathway
flux_matrix <- as.matrix(flux)
pathways <- unique(pathway_map)
pathway_flux <- sapply(pathways, function(pw) {
rxns <- names(pathway_map)[pathway_map == pw]
rxns <- intersect(rxns, rownames(flux_matrix))
if (length(rxns) > 0) {
mean(abs(flux_matrix[rxns, ]), na.rm = TRUE)
} else {
NA
}
})
# Remove NA and get top 15
pathway_flux <- sort(pathway_flux[!is.na(pathway_flux)], decreasing = TRUE)
top_pathways <- head(pathway_flux, 15)
# Create data frame
pathway_df <- data.frame(
Pathway = factor(names(top_pathways), levels = rev(names(top_pathways))),
Activity = top_pathways
)
# Plot
ggplot(pathway_df, aes(x = Activity, y = Pathway)) +
geom_bar(stat = "identity", fill = "#2c3e50", width = 0.7) +
labs(
title = "Top 15 Active Metabolic Pathways",
subtitle = "Based on mean absolute flux",
x = "Mean Absolute Flux",
y = ""
) +
theme_minimal() +
theme(
plot.title = element_text(hjust = 0.5, size = 14, face = "bold"),
plot.subtitle = element_text(hjust = 0.5, size = 10, color = "gray50"),
axis.text.y = element_text(size = 9)
)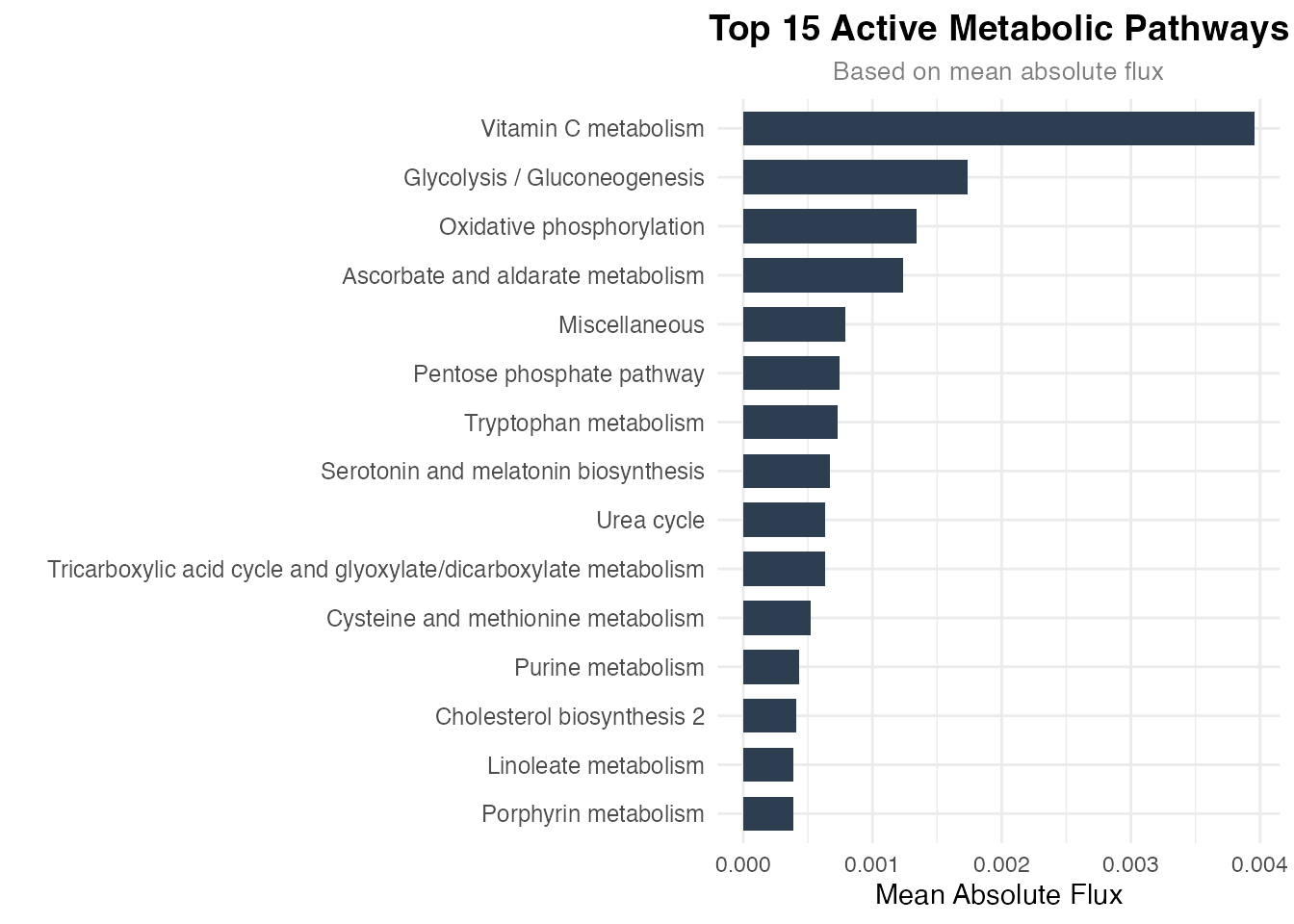
Mean absolute flux by metabolic pathway (top 15)
Visualization 5: Sample Correlation
# Calculate correlation
cor_matrix <- cor(as.matrix(flux), use = "pairwise.complete.obs")
# Convert to long format
cor_df <- data.frame(
Sample1 = rep(rownames(cor_matrix), ncol(cor_matrix)),
Sample2 = rep(colnames(cor_matrix), each = nrow(cor_matrix)),
Correlation = as.vector(cor_matrix)
)
# Plot
ggplot(cor_df, aes(x = Sample1, y = Sample2, fill = Correlation)) +
geom_tile() +
geom_text(aes(label = round(Correlation, 2)), size = 4) +
scale_fill_gradient2(
low = "#2166ac", mid = "white", high = "#b2182b",
midpoint = 0.5, limits = c(0, 1)
) +
labs(
title = "Sample Correlation Matrix",
subtitle = "Based on metabolic flux profiles",
x = "", y = ""
) +
theme_minimal() +
theme(
plot.title = element_text(hjust = 0.5, size = 14, face = "bold"),
plot.subtitle = element_text(hjust = 0.5, size = 10, color = "gray50"),
axis.text.x = element_text(angle = 45, hjust = 1)
)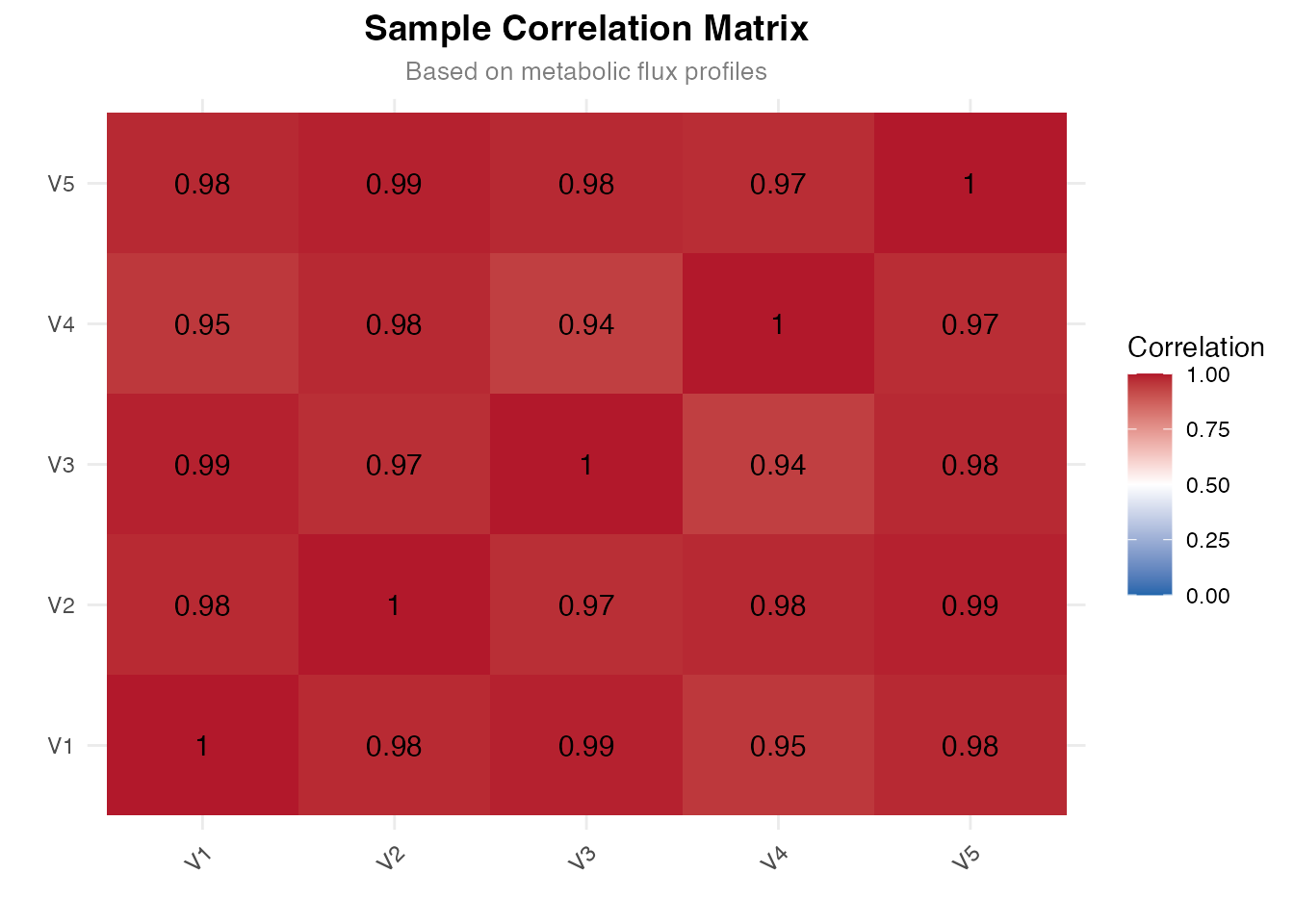
Correlation matrix of metabolic flux profiles
Visualization 6: Exchange Reaction Analysis
# Identify exchange reactions
exchange_idx <- grep("Exchange", human_gem$SUBSYSTEM)
exchange_rxns <- human_gem$ID[exchange_idx]
exchange_rxns <- intersect(exchange_rxns, rownames(flux))
# Get mean flux
exchange_flux <- rowMeans(flux[exchange_rxns, ])
# Classify
exchange_df <- data.frame(
Reaction = names(exchange_flux),
Flux = exchange_flux,
Type = ifelse(exchange_flux < -0.001, "Uptake",
ifelse(exchange_flux > 0.001, "Secretion", "Inactive"))
)
# Summary counts
type_counts <- table(exchange_df$Type)
cat("\nExchange reaction summary:\n")
#>
#> Exchange reaction summary:
print(type_counts)
#>
#> Inactive Secretion Uptake
#> 1598 27 23
# Plot distribution
ggplot(exchange_df[exchange_df$Type != "Inactive", ],
aes(x = Flux, fill = Type)) +
geom_histogram(bins = 50, alpha = 0.7, position = "identity") +
scale_fill_manual(values = c("Uptake" = "#e74c3c", "Secretion" = "#27ae60")) +
labs(
title = "Exchange Reaction Distribution",
subtitle = sprintf("Uptake: %d, Secretion: %d, Inactive: %d",
type_counts["Uptake"], type_counts["Secretion"],
type_counts["Inactive"]),
x = "Mean Flux",
y = "Count"
) +
theme_minimal() +
theme(
plot.title = element_text(hjust = 0.5, size = 14, face = "bold"),
plot.subtitle = element_text(hjust = 0.5, size = 10, color = "gray50")
)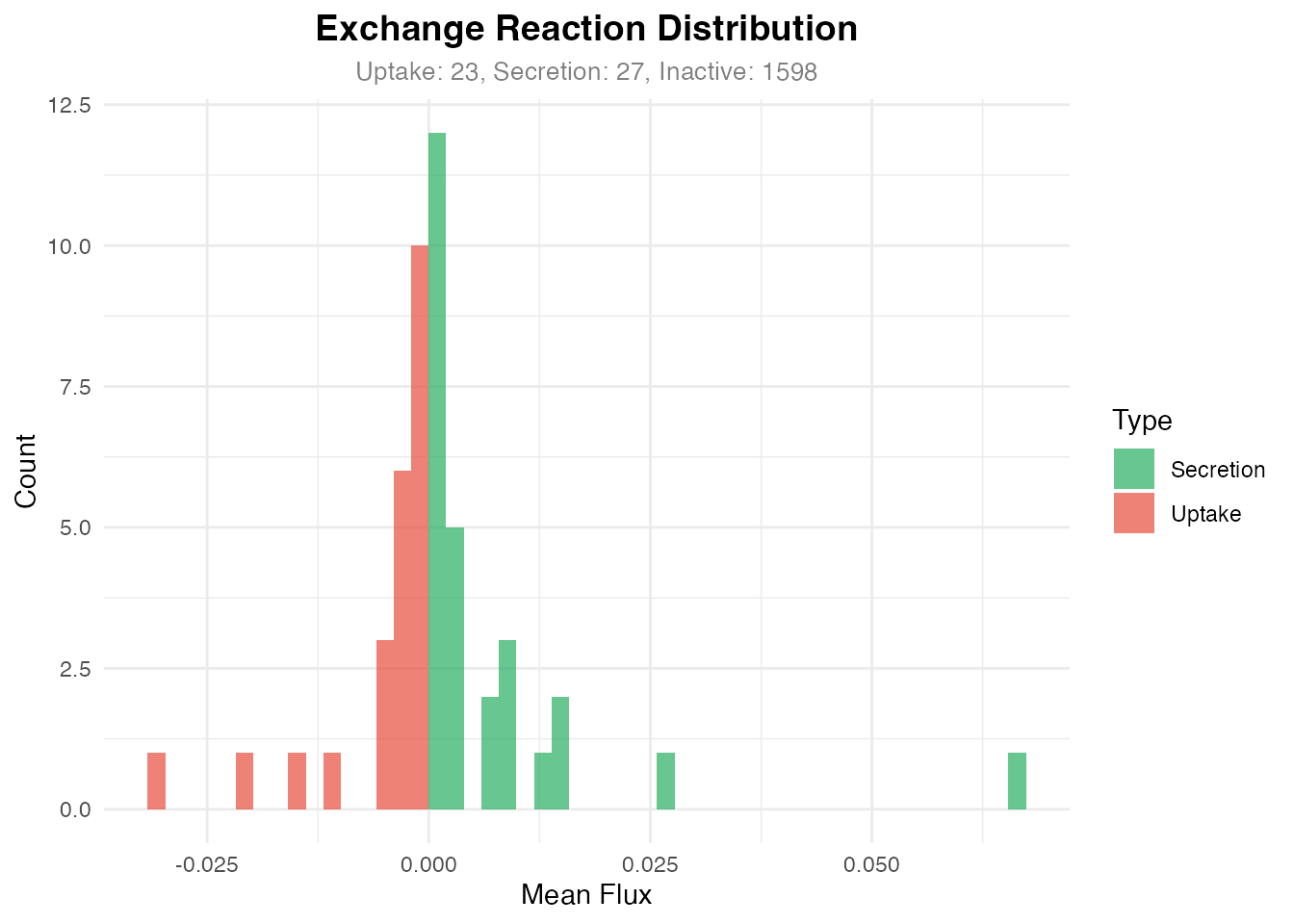
Distribution of uptake vs secretion reactions
Summary Statistics
cat("========================================\n")
#> ========================================
cat("METAFLUX Analysis Summary\n")
#> METAFLUX Analysis Summary
cat("========================================\n\n")
#> ========================================
cat("Input Data:\n")
#> Input Data:
cat(sprintf(" - Genes: %d\n", nrow(bulk_test_example)))
#> - Genes: 58581
cat(sprintf(" - Samples: %d\n", ncol(bulk_test_example)))
#> - Samples: 5
cat("\nOutput:\n")
#>
#> Output:
cat(sprintf(" - Reactions analyzed: %d\n", nrow(flux)))
#> - Reactions analyzed: 13082
cat(sprintf(" - Flux range: [%.4f, %.4f]\n", min(flux), max(flux)))
#> - Flux range: [-0.0840, 0.0841]
cat("\nKey Metabolites (mean flux):\n")
#>
#> Key Metabolites (mean flux):
for (met in names(key_rxns)) {
val <- mean(flux[key_rxns[met], ])
cat(sprintf(" - %s: %.4f\n", met, val))
}
#> - Glucose: -0.0052
#> - Lactate: 0.0020
#> - Glutamine: -0.0015
#> - Pyruvate: 0.0038Session Information
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggplot2_4.0.1 METAFLUX_2.2.0
#>
#> loaded via a namespace (and not attached):
#> [1] deldir_2.0-4 pbapply_1.7-4 gridExtra_2.3
#> [4] rlang_1.1.7 magrittr_2.0.4 RcppAnnoy_0.0.23
#> [7] otel_0.2.0 spatstat.geom_3.6-1 matrixStats_1.5.0
#> [10] ggridges_0.5.7 compiler_4.4.0 png_0.1-8
#> [13] systemfonts_1.3.1 vctrs_0.7.0 reshape2_1.4.5
#> [16] stringr_1.6.0 pkgconfig_2.0.3 fastmap_1.2.0
#> [19] labeling_0.4.3 promises_1.5.0 rmarkdown_2.30
#> [22] ragg_1.5.0 purrr_1.2.1 xfun_0.56
#> [25] cachem_1.1.0 jsonlite_2.0.0 goftest_1.2-3
#> [28] later_1.4.5 spatstat.utils_3.2-1 irlba_2.3.5.1
#> [31] parallel_4.4.0 cluster_2.1.8.1 R6_2.6.1
#> [34] ica_1.0-3 spatstat.data_3.1-9 bslib_0.9.0
#> [37] stringi_1.8.7 RColorBrewer_1.1-3 reticulate_1.44.1
#> [40] spatstat.univar_3.1-6 parallelly_1.46.1 lmtest_0.9-40
#> [43] jquerylib_0.1.4 scattermore_1.2 iterators_1.0.14
#> [46] Rcpp_1.1.1 knitr_1.51 tensor_1.5.1
#> [49] future.apply_1.20.1 zoo_1.8-15 sctransform_0.4.3
#> [52] httpuv_1.6.16 Matrix_1.7-4 splines_4.4.0
#> [55] igraph_2.2.1 tidyselect_1.2.1 abind_1.4-8
#> [58] dichromat_2.0-0.1 yaml_2.3.12 doParallel_1.0.17
#> [61] spatstat.random_3.4-3 spatstat.explore_3.6-0 codetools_0.2-20
#> [64] miniUI_0.1.2 listenv_0.10.0 plyr_1.8.9
#> [67] lattice_0.22-7 tibble_3.3.1 withr_3.0.2
#> [70] shiny_1.12.1 S7_0.2.1 ROCR_1.0-11
#> [73] evaluate_1.0.5 Rtsne_0.17 future_1.69.0
#> [76] desc_1.4.3 survival_3.8-3 polyclip_1.10-7
#> [79] fitdistrplus_1.2-4 osqp_0.6.3.3 pillar_1.11.1
#> [82] Seurat_4.4.0 KernSmooth_2.23-26 foreach_1.5.2
#> [85] plotly_4.11.0 generics_0.1.4 sp_2.2-0
#> [88] scales_1.4.0 globals_0.18.0 xtable_1.8-4
#> [91] glue_1.8.0 lazyeval_0.2.2 tools_4.4.0
#> [94] data.table_1.18.0 RANN_2.6.2 fs_1.6.6
#> [97] leiden_0.4.3.1 cowplot_1.2.0 grid_4.4.0
#> [100] tidyr_1.3.2 nlme_3.1-168 patchwork_1.3.2
#> [103] cli_3.6.5 spatstat.sparse_3.1-0 textshaping_1.0.4
#> [106] viridisLite_0.4.2 dplyr_1.1.4 uwot_0.2.4
#> [109] gtable_0.3.6 sass_0.4.10 digest_0.6.39
#> [112] progressr_0.18.0 ggrepel_0.9.6 htmlwidgets_1.6.4
#> [115] SeuratObject_4.1.4 farver_2.1.2 htmltools_0.5.9
#> [118] pkgdown_2.1.3 lifecycle_1.0.5 httr_1.4.7
#> [121] mime_0.13 MASS_7.3-65