SVG: A Comprehensive R Package for Spatially Variable Gene Detection
Zaoqu Liu
2026-01-23
Source:vignettes/SVG-introduction.Rmd
SVG-introduction.RmdAbstract
Spatial transcriptomics has emerged as a transformative technology in
modern biology, enabling the simultaneous measurement of gene expression
and spatial localization within tissues. A fundamental analytical
challenge is the identification of Spatially Variable Genes
(SVGs) — genes exhibiting non-random spatial expression
patterns that may reflect underlying biological structures or gradients.
This vignette provides a comprehensive introduction to the
SVG package, which implements six state-of-the-art SVG
detection methods within a unified computational framework. We present
the mathematical foundations of each method, demonstrate their
application through extensive examples, compare their performance
characteristics, and provide practical guidelines for method selection
in various experimental contexts.
Keywords: Spatial transcriptomics, Spatially variable genes, Moran’s I, Gaussian processes, Spatial statistics, R package
Introduction
Background and Motivation
The emergence of spatial transcriptomics technologies, including 10x Genomics Visium, Slide-seq, MERFISH, and seqFISH, has revolutionized our understanding of tissue architecture by preserving the spatial context of gene expression measurements. Unlike single-cell RNA sequencing, which dissociates cells from their native environment, spatial transcriptomics maintains the positional information of transcripts, enabling the study of:
- Tissue organization and cellular neighborhoods
- Spatial gradients in developmental processes
- Cell-cell communication and signaling microenvironments
- Pathological patterns in disease contexts
A critical first step in spatial transcriptomics analysis is identifying genes whose expression patterns exhibit significant spatial structure — the so-called Spatially Variable Genes (SVGs). These genes serve as:
- Markers for distinct tissue regions or cell populations
- Candidates for spatial signaling pathways
- Features for downstream clustering and trajectory analysis
Package Overview
The SVG package provides a unified, high-performance
implementation of multiple SVG detection methods:
| Method | Statistical Framework | Key Features |
|---|---|---|
| MERINGUE | Moran’s I with spatial networks | Network-based, interpretable statistic |
| binSpect | Fisher’s exact test | Fast, robust to outliers |
| SPARK-X | Kernel-based association | Multi-scale pattern detection |
| Seurat | Moran’s I with distance weights | Compatible with Seurat workflow |
| nnSVG | Gaussian processes (NNGP) | Full statistical model, effect size |
| MarkVario | Mark variogram | Non-parametric spatial statistics |
Each method has distinct theoretical foundations and practical trade-offs, which we systematically explore in this vignette.
Mathematical Foundations
Understanding the mathematical principles underlying SVG detection methods is essential for appropriate method selection and result interpretation. This section provides rigorous derivations of the key statistical frameworks.
Spatial Autocorrelation: Moran’s I Statistic
Definition and Intuition
Moran’s I is a classical measure of global spatial autocorrelation, introduced by Patrick Moran in 1950. For gene expression values measured at spatial locations with coordinates , Moran’s I is defined as:
where:
- is the spatial weight between locations and
- is the sample mean
- is the spatial weights matrix
Interpretation:
- : Strong positive autocorrelation (similar values cluster)
- : Strong negative autocorrelation (dissimilar values neighbor each other)
- : Random spatial pattern
Statistical Inference
Under the null hypothesis of complete spatial randomness (CSR), the expected value of Moran’s I is:
The variance under the randomization assumption is given by:
where:
- (sum of all weights)
The standardized test statistic follows an asymptotic normal distribution:
Spatial Weights Specifications
The choice of spatial weights matrix is crucial and method-specific:
Binary Adjacency (MERINGUE):
Neighbors can be defined via: - Delaunay triangulation: Natural neighborhood based on Voronoi tessellation - K-nearest neighbors (KNN): Fixed number of closest points
Inverse Distance (Seurat):
where is the Euclidean distance.
Gaussian Kernel:
where is the bandwidth parameter.
Kernel-Based Association Tests: SPARK-X
Variance Component Score Test
SPARK-X employs a kernel-based framework to detect spatial patterns through variance component testing. For centered gene expression , the test statistic is:
where is a kernel matrix capturing spatial similarity.
Multiple Kernel Types
SPARK-X employs three classes of kernels to detect diverse spatial patterns:
1. Linear (Projection) Kernel:
Detects linear gradients and directional trends.
2. Gaussian RBF Kernels (5 scales):
where bandwidths correspond to the 20th, 40th, 60th, 80th, and 100th percentiles of pairwise distances. These detect localized hotspots of varying sizes.
3. Cosine/Periodic Kernels (5 scales):
Detect periodic or oscillating spatial patterns.
P-value Computation and Combination
For each kernel, p-values are computed using Davies’ method for quadratic forms in normal variables. The distribution of under the null follows:
where are eigenvalues of the centered kernel matrix.
P-values from all 11 kernels are combined using the Aggregated Cauchy Association Test (ACAT):
The combined p-value is then:
ACAT is robust to dependency among tests and maintains correct type I error.
Binary Spatial Enrichment: binSpect
Methodology
The binSpect method from Giotto converts continuous expression into binary categories and tests for spatial enrichment using contingency tables.
Step 1: Expression Binarization
For gene , expression values are classified as “high” or “low” using k-means clustering ():
Step 2: Spatial Contingency Table
For all pairs of neighboring spots in the spatial network:
| Neighbor High | Neighbor Low | |
|---|---|---|
| Spot High | ||
| Spot Low |
Step 3: Fisher’s Exact Test
The odds ratio quantifies spatial enrichment:
- : High-expressing cells cluster together
- : High-expressing cells avoid each other
- : Random spatial distribution
Fisher’s exact test provides exact p-values for the null hypothesis .
Nearest-Neighbor Gaussian Processes: nnSVG
Full Statistical Model
nnSVG provides the most complete statistical framework by modeling spatial correlation through Gaussian processes:
where:
- is the expression vector
- is the design matrix (covariates)
- is the coefficient vector
- is the spatial random effect
- is the nugget (non-spatial variance)
Covariance Function
The spatial covariance is typically modeled using the exponential function:
where is the range parameter controlling spatial decay.
Installation and Setup
# Install from GitHub
devtools::install_github("Zaoqu-Liu/SVG")
# Install all optional dependencies
install.packages(c("geometry", "RANN", "BRISC", "CompQuadForm",
"spatstat.geom", "spatstat.explore"))
# Load the SVG package
library(SVG)
# Load the built-in example dataset
data(example_svg_data)
# Display dataset structure
cat("Dataset components:\n")
#> Dataset components:
names(example_svg_data)
#> [1] "counts" "logcounts" "spatial_coords" "gene_info"
#> [5] "params"Data Description and Visualization
Simulated Spatial Transcriptomics Data
The example_svg_data contains simulated spatial
transcriptomics data designed to benchmark SVG detection methods:
# Extract components
expr_counts <- example_svg_data$counts # Raw counts
expr_log <- example_svg_data$logcounts # Log-normalized
coords <- example_svg_data$spatial_coords # Spatial coordinates
gene_info <- example_svg_data$gene_info # Gene metadata
cat("Data Dimensions:\n")
#> Data Dimensions:
cat(" Number of spots:", ncol(expr_counts), "\n")
#> Number of spots: 500
cat(" Number of genes:", nrow(expr_counts), "\n")
#> Number of genes: 200
cat(" True SVGs:", sum(gene_info$is_svg), "\n")
#> True SVGs: 50
cat(" Non-SVGs:", sum(!gene_info$is_svg), "\n")
#> Non-SVGs: 150
cat("\nSpatial Pattern Types:\n")
#>
#> Spatial Pattern Types:
print(table(gene_info$pattern_type))
#>
#> cluster gradient hotspot none periodic
#> 11 13 13 150 13Spatial Spot Layout
The spots are arranged in a hexagonal grid pattern, mimicking the 10x Genomics Visium platform:
par(mar = c(4, 4, 3, 2))
plot(coords[, 1], coords[, 2],
pch = 19, cex = 0.9,
col = adjustcolor("steelblue", alpha.f = 0.7),
xlab = "X coordinate (μm)",
ylab = "Y coordinate (μm)",
main = "Spatial Spot Layout (Hexagonal Grid)",
asp = 1)
grid(lty = 2, col = "gray80")
# Add spot count annotation
mtext(paste("n =", nrow(coords), "spots"), side = 3, line = 0.3, cex = 0.9)
Gene Expression Distribution
par(mfrow = c(1, 2), mar = c(4, 4, 3, 1))
# Raw counts distribution
hist(log10(rowMeans(expr_counts) + 1), breaks = 30,
col = "lightblue", border = "white",
xlab = expression(log[10](Mean~Count + 1)),
ylab = "Number of Genes",
main = "Gene Expression Distribution")
# Spot library size
hist(log10(colSums(expr_counts)), breaks = 30,
col = "lightgreen", border = "white",
xlab = expression(log[10](Library~Size)),
ylab = "Number of Spots",
main = "Spot Library Size Distribution")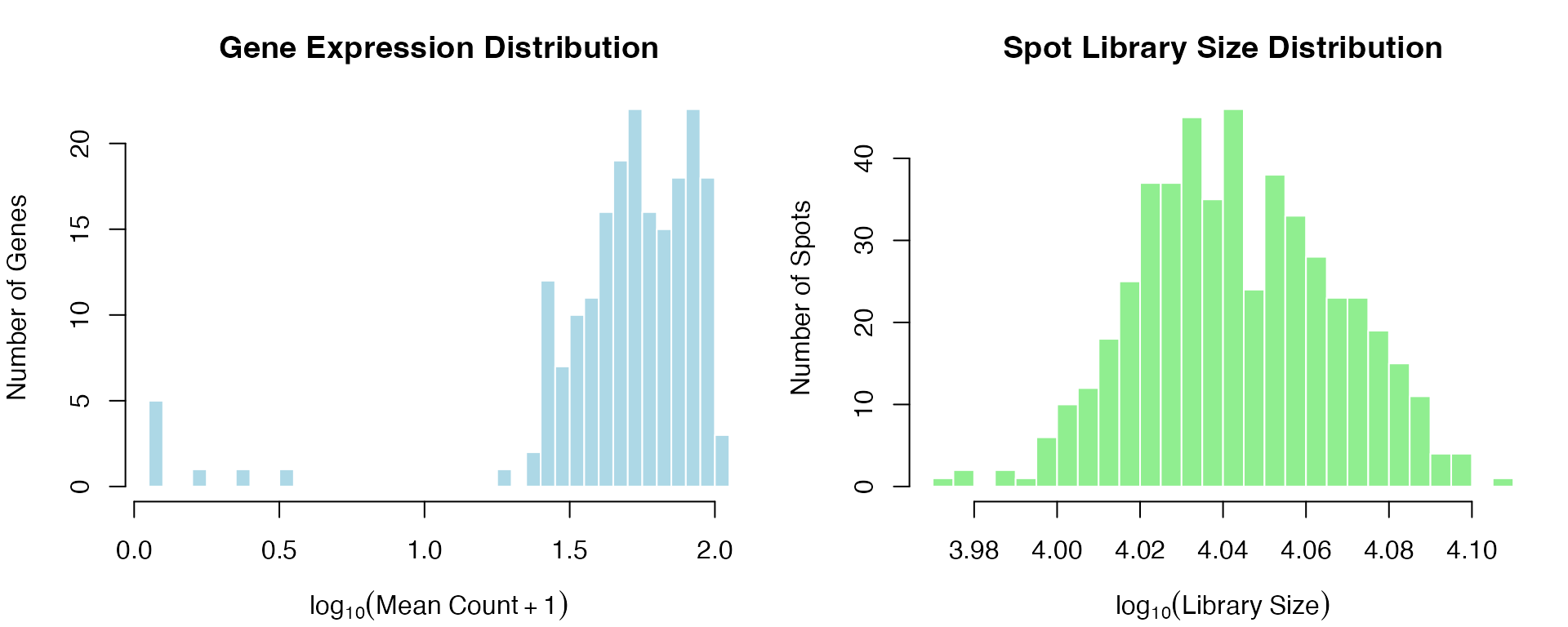
Spatial Expression Pattern Visualization
The simulated data contains four distinct spatial pattern types for SVGs:
# Define color palette for expression visualization
expr_colors <- function(x, pal = "RdYlBu") {
x_scaled <- (x - min(x, na.rm = TRUE)) / (max(x, na.rm = TRUE) - min(x, na.rm = TRUE) + 1e-10)
if (pal == "RdYlBu") {
colors <- colorRampPalette(c("#313695", "#4575B4", "#74ADD1", "#ABD9E9",
"#E0F3F8", "#FFFFBF", "#FEE090", "#FDAE61",
"#F46D43", "#D73027", "#A50026"))(100)
} else {
colors <- colorRampPalette(c("navy", "white", "firebrick3"))(100)
}
colors[pmax(1, ceiling(x_scaled * 99) + 1)]
}
# Get example genes for each pattern type
pattern_types <- c("gradient", "hotspot", "periodic", "cluster")
pattern_genes <- sapply(pattern_types, function(pt) {
which(gene_info$pattern_type == pt)[1]
})
# Get non-SVG example
non_svg_gene <- which(!gene_info$is_svg)[1]
par(mfrow = c(2, 3), mar = c(4, 4, 3, 1))
# Plot each pattern type
for (i in seq_along(pattern_types)) {
gene_idx <- pattern_genes[i]
gene_name <- rownames(expr_log)[gene_idx]
gene_expr <- expr_log[gene_idx, ]
plot(coords[, 1], coords[, 2],
pch = 19, cex = 1.3,
col = expr_colors(gene_expr),
xlab = "X", ylab = "Y",
main = paste0(gene_name, "\n(", pattern_types[i], " pattern)"),
asp = 1)
}
# Plot non-SVG
gene_expr <- expr_log[non_svg_gene, ]
plot(coords[, 1], coords[, 2],
pch = 19, cex = 1.3,
col = expr_colors(gene_expr),
xlab = "X", ylab = "Y",
main = paste0(rownames(expr_log)[non_svg_gene], "\n(non-SVG, random)"),
asp = 1)
# Add color legend
plot.new()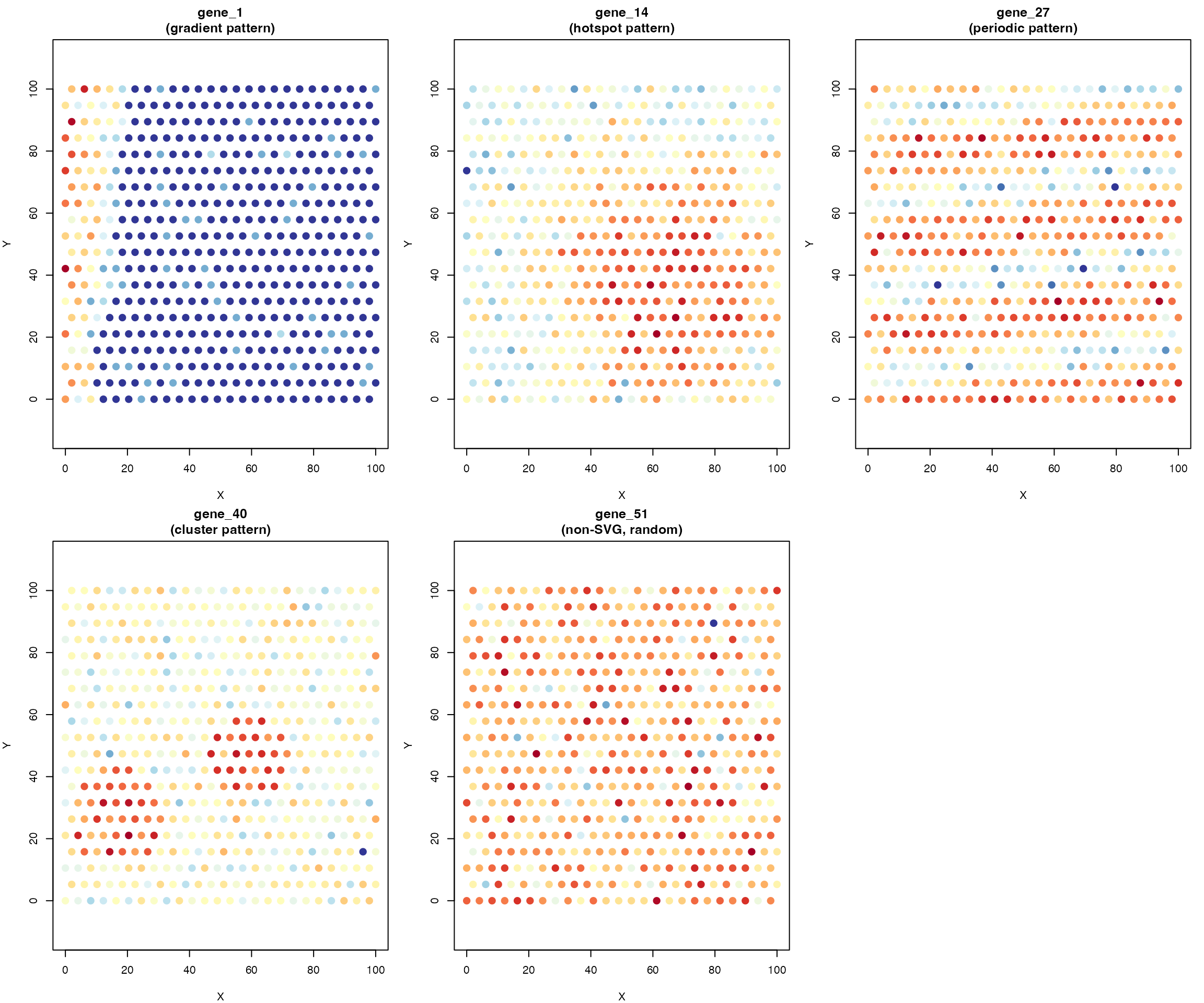
par(mar = c(1, 1, 1, 1))
legend_colors <- colorRampPalette(c("#313695", "#FFFFBF", "#A50026"))(100)
legend_image <- as.raster(matrix(rev(legend_colors), ncol = 1))
plot(c(0, 1), c(0, 1), type = "n", axes = FALSE, xlab = "", ylab = "")
text(0.5, 0.95, "Expression Level", cex = 1.2, font = 2)
rasterImage(legend_image, 0.3, 0.1, 0.7, 0.85)
text(0.75, 0.1, "Low", cex = 0.9)
text(0.75, 0.85, "High", cex = 0.9)
SVG Detection: Method-by-Method Tutorial
Method 1: MERINGUE (Moran’s I with Spatial Networks)
Algorithm Overview
MERINGUE computes Moran’s I statistic using a sparse spatial adjacency network, offering computational efficiency while maintaining statistical power.
Key Steps:
- Construct spatial neighborhood network (Delaunay or KNN)
- Compute Moran’s I for each gene
- Calculate analytical p-values under normal approximation
- Apply multiple testing correction
Running MERINGUE
# Run MERINGUE with KNN network
results_meringue <- CalSVG_MERINGUE(
expr_matrix = expr_log,
spatial_coords = coords,
network_method = "knn", # Network construction method
k = 10, # Number of neighbors
alternative = "greater", # Test for positive autocorrelation
adjust_method = "BH", # Benjamini-Hochberg correction
verbose = FALSE
)
# Display top SVGs
cat("Top 10 SVGs by MERINGUE:\n")
#> Top 10 SVGs by MERINGUE:
head(results_meringue[, c("gene", "observed", "expected", "z_score", "p.value", "p.adj")], 10)
#> gene observed expected z_score p.value p.adj
#> 1 gene_1 0.7667350 -0.002004008 39.717880 0 0
#> 2 gene_2 0.4929258 -0.002004008 25.686514 0 0
#> 3 gene_3 0.1744444 -0.002004008 9.287831 0 0
#> 4 gene_4 0.4593775 -0.002004008 23.759281 0 0
#> 5 gene_5 0.7236521 -0.002004008 37.422043 0 0
#> 6 gene_6 0.1587466 -0.002004008 8.496923 0 0
#> 7 gene_7 0.9056749 -0.002004008 46.670022 0 0
#> 8 gene_8 0.2925418 -0.002004008 15.388682 0 0
#> 9 gene_9 0.8262118 -0.002004008 42.721016 0 0
#> 10 gene_10 0.4378317 -0.002004008 22.658627 0 0Visualizing MERINGUE Results
par(mfrow = c(1, 2), mar = c(4, 4, 3, 1))
# Moran's I distribution
hist(results_meringue$observed, breaks = 40,
col = "steelblue", border = "white",
xlab = "Moran's I",
ylab = "Number of Genes",
main = "Distribution of Moran's I Statistics")
abline(v = results_meringue$expected[1], col = "red", lwd = 2, lty = 2)
legend("topright", legend = "E[I] under null", col = "red", lty = 2, lwd = 2)
# P-value distribution
hist(results_meringue$p.value, breaks = 40,
col = "coral", border = "white",
xlab = "P-value",
ylab = "Number of Genes",
main = "P-value Distribution")
abline(v = 0.05, col = "darkred", lwd = 2, lty = 2)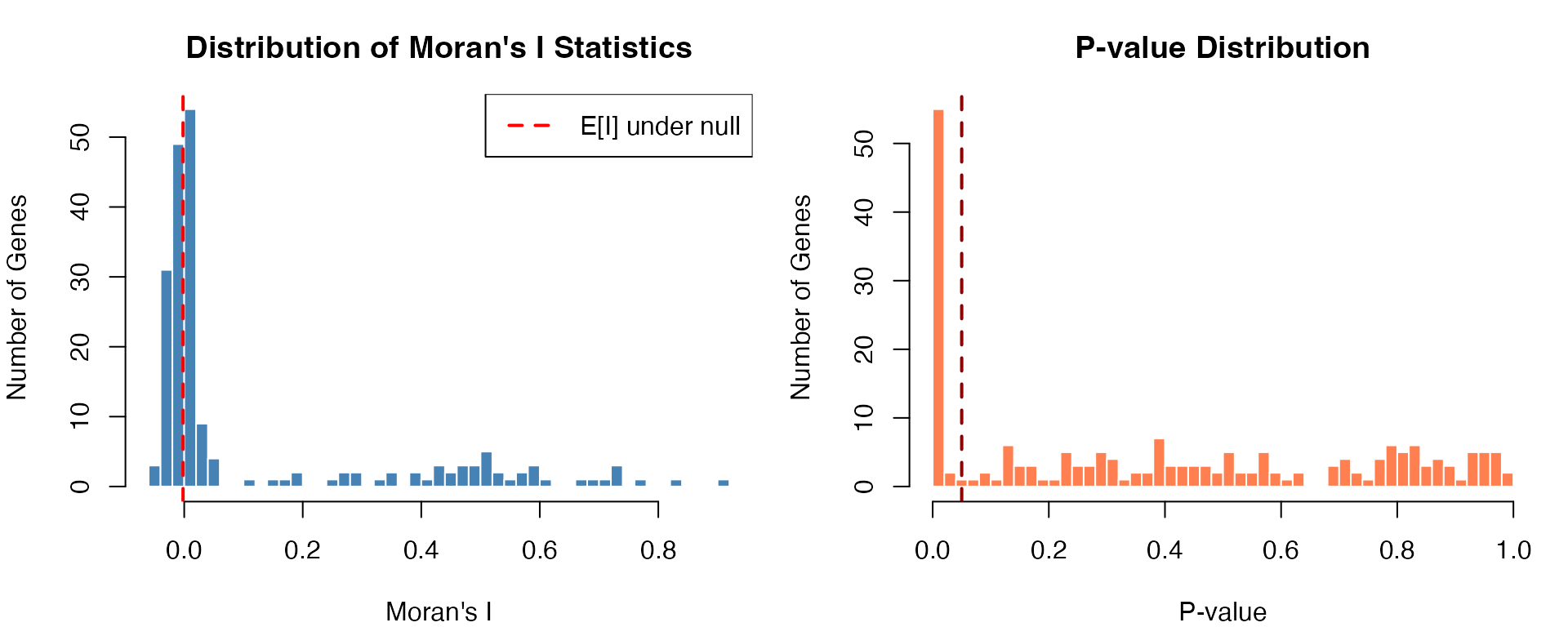
Method 2: binSpect (Binary Spatial Enrichment)
Algorithm Overview
binSpect converts continuous expression to binary categories and uses Fisher’s exact test to detect spatial clustering.
Key Steps:
- Binarize gene expression (k-means or rank-based)
- Build spatial neighborhood network
- Construct contingency tables for neighbor pairs
- Perform Fisher’s exact test for each gene
Running binSpect
# Run binSpect with k-means binarization
results_binspect <- CalSVG_binSpect(
expr_matrix = expr_log,
spatial_coords = coords,
bin_method = "kmeans", # Binarization method
network_method = "delaunay", # Network construction
do_fisher_test = TRUE, # Perform Fisher's test
adjust_method = "fdr", # FDR correction
verbose = FALSE
)
# Display top SVGs
cat("Top 10 SVGs by binSpect:\n")
#> Top 10 SVGs by binSpect:
head(results_binspect[, c("gene", "estimate", "p.value", "p.adj", "score")], 10)
#> gene estimate p.value p.adj score
#> 1 gene_7 239.82358 1.881408e-272 3.762816e-270 65166.186
#> 2 gene_9 352.47489 9.696440e-174 9.696440e-172 60982.874
#> 3 gene_1 320.18186 7.426544e-152 3.713272e-150 48388.833
#> 4 gene_41 102.16057 4.940292e-142 1.646764e-140 14435.927
#> 5 gene_42 50.21381 5.153274e-143 2.061310e-141 7144.818
#> 6 gene_47 68.45425 6.040472e-98 7.106438e-97 6655.049
#> 7 gene_19 30.19604 1.958767e-156 1.305845e-154 4701.766
#> 8 gene_46 33.49625 6.175115e-138 1.543779e-136 4595.999
#> 9 gene_2 73.73039 6.739613e-61 4.212258e-60 4436.458
#> 10 gene_48 33.05526 4.694890e-123 9.389780e-122 4043.597Visualizing binSpect Results
par(mfrow = c(1, 2), mar = c(4, 4, 3, 1))
# Odds ratio distribution
or_log <- log2(results_binspect$estimate + 0.01)
hist(or_log, breaks = 40,
col = "mediumpurple", border = "white",
xlab = expression(log[2](Odds~Ratio)),
ylab = "Number of Genes",
main = "Distribution of Odds Ratios")
abline(v = 0, col = "red", lwd = 2, lty = 2)
# Score distribution (combines p-value and OR)
hist(results_binspect$score, breaks = 40,
col = "darkorange", border = "white",
xlab = "binSpect Score",
ylab = "Number of Genes",
main = "Distribution of binSpect Scores")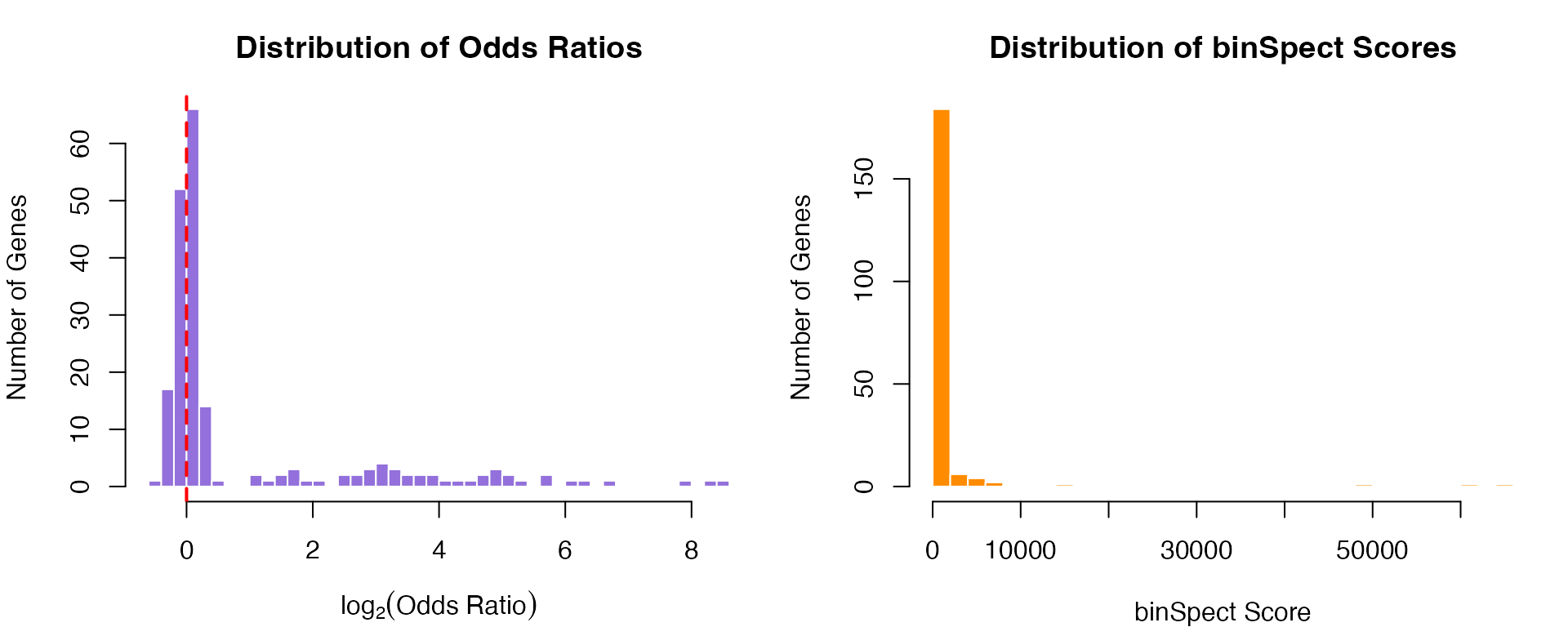
Method 3: SPARK-X (Kernel-Based Association)
Algorithm Overview
SPARK-X uses multiple spatial kernels to detect diverse pattern types, combining results via ACAT.
Kernels Used:
- 1 linear (projection) kernel
- 5 Gaussian RBF kernels (varying bandwidths)
- 5 cosine/periodic kernels (varying frequencies)
Running SPARK-X
# Run SPARK-X with mixture of kernels
# Note: Uses raw counts (not log-transformed)
results_sparkx <- CalSVG_SPARKX(
expr_matrix = expr_counts, # Raw counts recommended
spatial_coords = coords,
kernel_option = "mixture", # All 11 kernels
adjust_method = "BY", # Benjamini-Yekutieli (conservative)
verbose = FALSE
)
# Display top SVGs
cat("Top 10 SVGs by SPARK-X:\n")
#> Top 10 SVGs by SPARK-X:
head(results_sparkx[, c("gene", "p.value", "p.adj")], 10)
#> gene p.value p.adj
#> 1 gene_4 8.282182e-179 9.736585e-176
#> 2 gene_10 9.435583e-163 5.546265e-160
#> 3 gene_14 6.294663e-160 2.466681e-157
#> 4 gene_12 2.134502e-158 6.273335e-156
#> 5 gene_5 2.592901e-154 6.096461e-152
#> 6 gene_18 7.617152e-144 1.492462e-141
#> 7 gene_15 2.459916e-134 4.131274e-132
#> 8 gene_25 3.889349e-134 5.715428e-132
#> 9 gene_22 5.640595e-134 7.367910e-132
#> 10 gene_17 3.852617e-130 4.529160e-128Visualizing SPARK-X Results
par(mfrow = c(1, 2), mar = c(4, 4, 3, 1))
# Combined p-value distribution
hist(-log10(results_sparkx$p.value + 1e-300), breaks = 40,
col = "seagreen", border = "white",
xlab = expression(-log[10](p-value)),
ylab = "Number of Genes",
main = "SPARK-X P-value Distribution")
# Volcano-style plot
pval_log <- -log10(results_sparkx$p.adj + 1e-300)
plot(seq_along(pval_log), pval_log,
pch = 19, cex = 0.6,
col = ifelse(pval_log > -log10(0.05), "red", "gray50"),
xlab = "Gene Index",
ylab = expression(-log[10](adjusted~p-value)),
main = "SPARK-X Significance Plot")
abline(h = -log10(0.05), col = "red", lty = 2)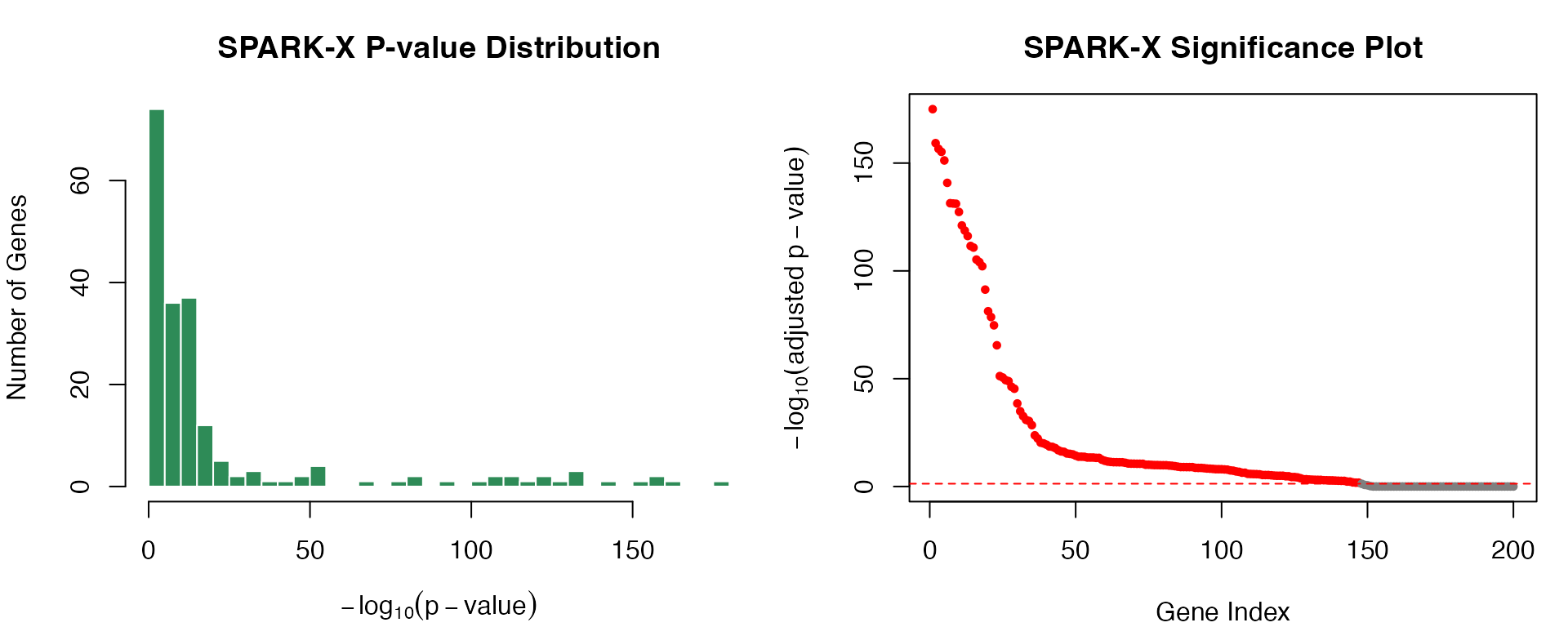
Method 4: Seurat (Moran’s I with Distance Weights)
Algorithm Overview
Seurat’s implementation uses inverse-squared distance weighting, emphasizing local spatial relationships.
Running Seurat Method
# Run Seurat Moran's I
results_seurat <- CalSVG_Seurat(
expr_matrix = expr_log,
spatial_coords = coords,
weight_scheme = "inverse_squared", # Seurat default
adjust_method = "BH",
verbose = FALSE
)
# Display top SVGs
cat("Top 10 SVGs by Seurat:\n")
#> Top 10 SVGs by Seurat:
head(results_seurat[, c("gene", "observed", "expected", "sd", "p.value", "p.adj")], 10)
#> gene observed expected sd p.value p.adj
#> 1 gene_7 0.6772728 -0.002004008 0.009605248 0 0
#> 2 gene_22 0.5457111 -0.002004008 0.009599242 0 0
#> 3 gene_17 0.5345832 -0.002004008 0.009597744 0 0
#> 4 gene_19 0.5149447 -0.002004008 0.009594016 0 0
#> 5 gene_9 0.5090348 -0.002004008 0.009574655 0 0
#> 6 gene_12 0.4738920 -0.002004008 0.009579223 0 0
#> 7 gene_25 0.4595807 -0.002004008 0.009597722 0 0
#> 8 gene_5 0.4564260 -0.002004008 0.009576880 0 0
#> 9 gene_14 0.4165174 -0.002004008 0.009595459 0 0
#> 10 gene_23 0.4098592 -0.002004008 0.009581536 0 0Unified Interface: CalSVG()
For convenience, all methods can be accessed through a single unified interface:
# Example: Run MERINGUE through unified interface
results_unified <- CalSVG(
expr_matrix = expr_log,
spatial_coords = coords,
method = "meringue", # Options: meringue, binspect, sparkx, seurat, nnsvg, markvario
network_method = "knn",
k = 10,
verbose = FALSE
)
cat("Unified interface results:\n")
#> Unified interface results:
head(results_unified[, c("gene", "p.value", "p.adj")], 5)
#> gene p.value p.adj
#> 1 gene_1 0 0
#> 2 gene_2 0 0
#> 3 gene_3 0 0
#> 4 gene_4 0 0
#> 5 gene_5 0 0Comprehensive Method Comparison
Performance Metrics
We evaluate all methods using standard classification metrics on the simulated data with known ground truth:
# Ground truth
truth <- gene_info$is_svg
# Function to calculate comprehensive metrics
calc_performance <- function(result, truth, pval_col = "p.adj", threshold = 0.05) {
detected <- result[[pval_col]] < threshold
detected[is.na(detected)] <- FALSE
TP <- sum(truth & detected)
FP <- sum(!truth & detected)
FN <- sum(truth & !detected)
TN <- sum(!truth & !detected)
list(
TP = TP, FP = FP, FN = FN, TN = TN,
Sensitivity = TP / (TP + FN),
Specificity = TN / (TN + FP),
Precision = TP / max(TP + FP, 1),
NPV = TN / max(TN + FN, 1),
F1 = 2 * TP / max(2 * TP + FP + FN, 1),
Accuracy = (TP + TN) / (TP + TN + FP + FN),
MCC = (TP * TN - FP * FN) / sqrt((TP + FP) * (TP + FN) * (TN + FP) * (TN + FN) + 1e-10)
)
}
# Calculate metrics for each method
metrics_list <- list(
MERINGUE = calc_performance(results_meringue, truth),
binSpect = calc_performance(results_binspect, truth),
`SPARK-X` = calc_performance(results_sparkx, truth),
Seurat = calc_performance(results_seurat, truth)
)
# Create metrics data frame
metrics_df <- do.call(rbind, lapply(names(metrics_list), function(m) {
data.frame(
Method = m,
Sensitivity = metrics_list[[m]]$Sensitivity,
Specificity = metrics_list[[m]]$Specificity,
Precision = metrics_list[[m]]$Precision,
F1 = metrics_list[[m]]$F1,
MCC = metrics_list[[m]]$MCC
)
}))
knitr::kable(metrics_df, digits = 3,
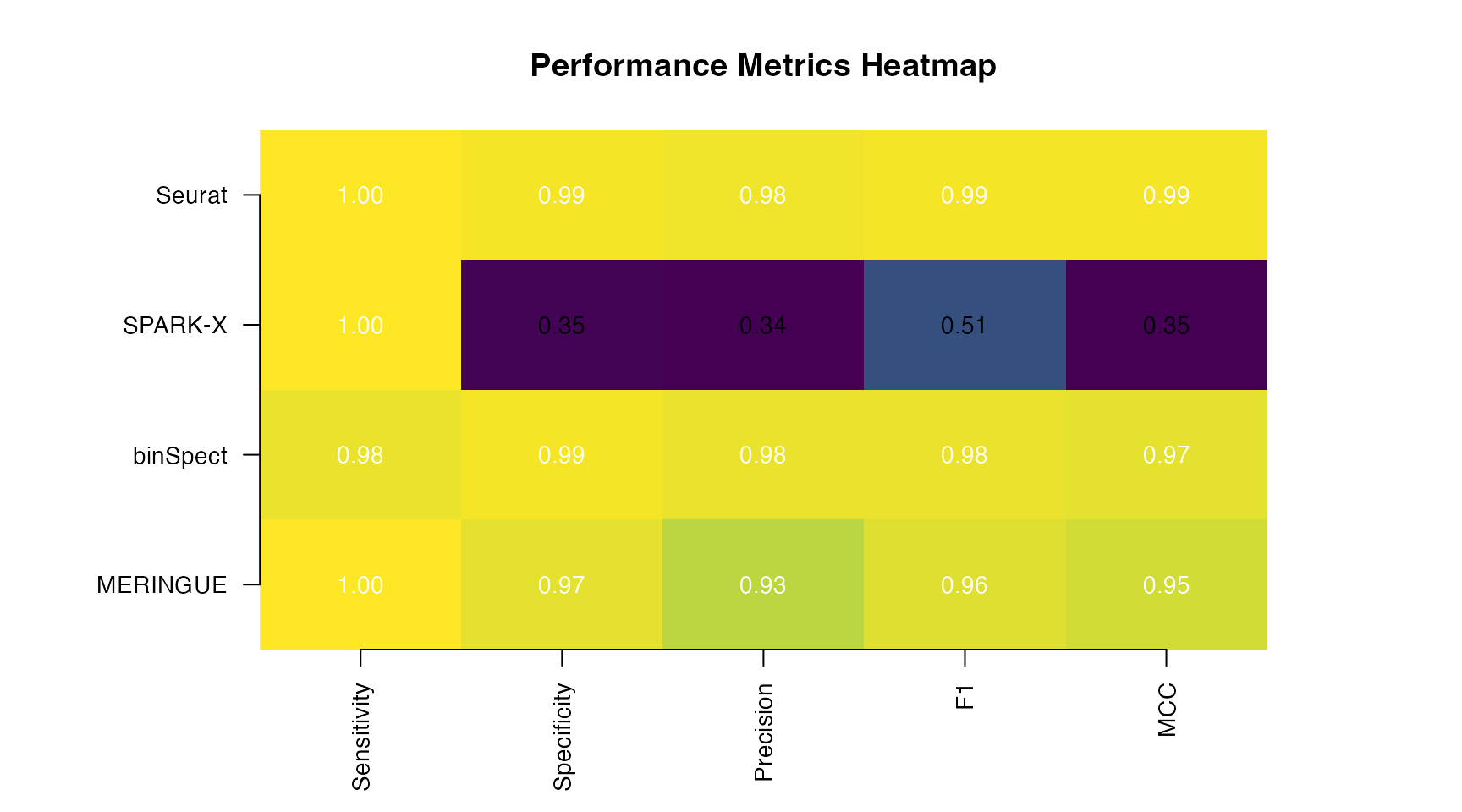
caption = "Performance Comparison on Simulated Data (FDR < 0.05)")| Method | Sensitivity | Specificity | Precision | F1 | MCC |
|---|---|---|---|---|---|
| MERINGUE | 1.00 | 0.973 | 0.926 | 0.962 | 0.949 |
| binSpect | 0.98 | 0.993 | 0.980 | 0.980 | 0.973 |
| SPARK-X | 1.00 | 0.353 | 0.340 | 0.508 | 0.347 |
| Seurat | 1.00 | 0.993 | 0.980 | 0.990 | 0.987 |
Visual Performance Comparison
# Prepare data for heatmap
metrics_matrix <- as.matrix(metrics_df[, -1])
rownames(metrics_matrix) <- metrics_df$Method
# Create heatmap
par(mar = c(5, 8, 4, 6))
image(t(metrics_matrix), axes = FALSE,
col = colorRampPalette(c("#440154", "#31688E", "#35B779", "#FDE725"))(100),
main = "Performance Metrics Heatmap")
axis(1, at = seq(0, 1, length.out = ncol(metrics_matrix)),
labels = colnames(metrics_matrix), las = 2, cex.axis = 0.9)
axis(2, at = seq(0, 1, length.out = nrow(metrics_matrix)),
labels = rownames(metrics_matrix), las = 1, cex.axis = 0.9)
# Add values
for (i in 1:nrow(metrics_matrix)) {
for (j in 1:ncol(metrics_matrix)) {
text((j - 1) / (ncol(metrics_matrix) - 1),
(i - 1) / (nrow(metrics_matrix) - 1),
sprintf("%.2f", metrics_matrix[i, j]),
cex = 0.9, col = ifelse(metrics_matrix[i, j] > 0.6, "white", "black"))
}
}
ROC Curve Analysis
# Function to compute ROC curve
compute_roc <- function(scores, truth, higher_is_better = FALSE) {
if (higher_is_better) {
scores <- -scores
}
scores[is.na(scores)] <- max(scores, na.rm = TRUE) + 1
thresholds <- sort(unique(c(-Inf, scores, Inf)))
tpr <- fpr <- numeric(length(thresholds))
for (i in seq_along(thresholds)) {
predicted <- scores <= thresholds[i]
tpr[i] <- sum(truth & predicted) / sum(truth)
fpr[i] <- sum(!truth & predicted) / sum(!truth)
}
# Calculate AUC using trapezoidal rule
ord <- order(fpr, tpr)
fpr <- fpr[ord]
tpr <- tpr[ord]
auc <- sum(diff(fpr) * (head(tpr, -1) + tail(tpr, -1)) / 2)
list(fpr = fpr, tpr = tpr, auc = auc)
}
# Compute ROC for each method
roc_meringue <- compute_roc(results_meringue$p.value, truth)
roc_binspect <- compute_roc(results_binspect$p.value, truth)
roc_sparkx <- compute_roc(results_sparkx$p.value, truth)
roc_seurat <- compute_roc(results_seurat$p.value, truth)
# Plot ROC curves
par(mar = c(5, 5, 4, 2))
plot(roc_meringue$fpr, roc_meringue$tpr, type = "l", lwd = 3, col = "#E41A1C",
xlab = "False Positive Rate (1 - Specificity)",
ylab = "True Positive Rate (Sensitivity)",
main = "Receiver Operating Characteristic (ROC) Curves",
xlim = c(0, 1), ylim = c(0, 1),
cex.lab = 1.2, cex.main = 1.3)
lines(roc_binspect$fpr, roc_binspect$tpr, lwd = 3, col = "#377EB8")
lines(roc_sparkx$fpr, roc_sparkx$tpr, lwd = 3, col = "#4DAF4A")
lines(roc_seurat$fpr, roc_seurat$tpr, lwd = 3, col = "#984EA3")
abline(0, 1, lty = 2, col = "gray50", lwd = 2)
# Add AUC values to legend
legend("bottomright",
legend = c(
sprintf("MERINGUE (AUC = %.3f)", roc_meringue$auc),
sprintf("binSpect (AUC = %.3f)", roc_binspect$auc),
sprintf("SPARK-X (AUC = %.3f)", roc_sparkx$auc),
sprintf("Seurat (AUC = %.3f)", roc_seurat$auc)
),
col = c("#E41A1C", "#377EB8", "#4DAF4A", "#984EA3"),
lwd = 3, cex = 1.0, bty = "n")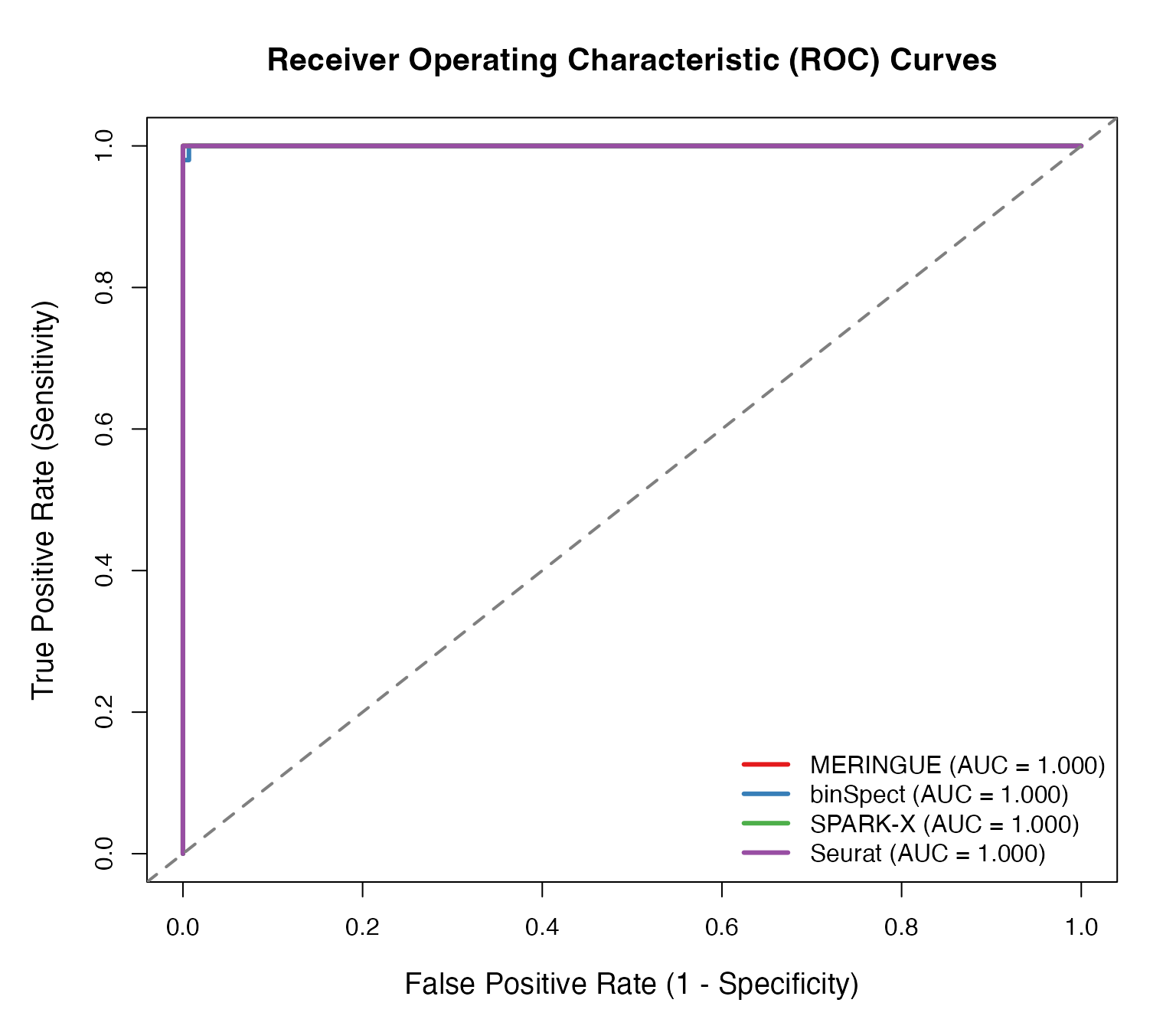
Overlap Analysis
# Get significant genes for each method
sig_genes <- list(
MERINGUE = results_meringue$gene[results_meringue$p.adj < 0.05],
binSpect = results_binspect$gene[results_binspect$p.adj < 0.05],
SPARKX = results_sparkx$gene[results_sparkx$p.adj < 0.05],
Seurat = results_seurat$gene[results_seurat$p.adj < 0.05]
)
# Calculate pairwise Jaccard indices
jaccard <- function(a, b) {
length(intersect(a, b)) / length(union(a, b))
}
methods <- names(sig_genes)
jaccard_matrix <- matrix(0, length(methods), length(methods))
rownames(jaccard_matrix) <- colnames(jaccard_matrix) <- methods
for (i in seq_along(methods)) {
for (j in seq_along(methods)) {
jaccard_matrix[i, j] <- jaccard(sig_genes[[i]], sig_genes[[j]])
}
}
# Visualize overlap
par(mfrow = c(1, 2), mar = c(5, 6, 4, 4))
# Jaccard similarity heatmap
image(jaccard_matrix, axes = FALSE,
col = colorRampPalette(c("white", "steelblue", "darkblue"))(100),
main = "Pairwise Jaccard Similarity")
axis(1, at = seq(0, 1, length.out = 4), labels = methods, las = 2, cex.axis = 0.9)
axis(2, at = seq(0, 1, length.out = 4), labels = methods, las = 1, cex.axis = 0.9)
for (i in 1:4) {
for (j in 1:4) {
text((j - 1) / 3, (i - 1) / 3,
sprintf("%.2f", jaccard_matrix[i, j]),
cex = 1.0, col = ifelse(jaccard_matrix[i, j] > 0.5, "white", "black"))
}
}
# Number of significant genes
barplot(sapply(sig_genes, length),
col = c("#E41A1C", "#377EB8", "#4DAF4A", "#984EA3"),
ylab = "Number of Significant Genes",
main = "Number of SVGs Detected",
las = 1, border = NA)
abline(h = sum(truth), lty = 2, col = "red", lwd = 2)
legend("topright", legend = "True SVGs (n=50)", lty = 2, col = "red", lwd = 2, bty = "n")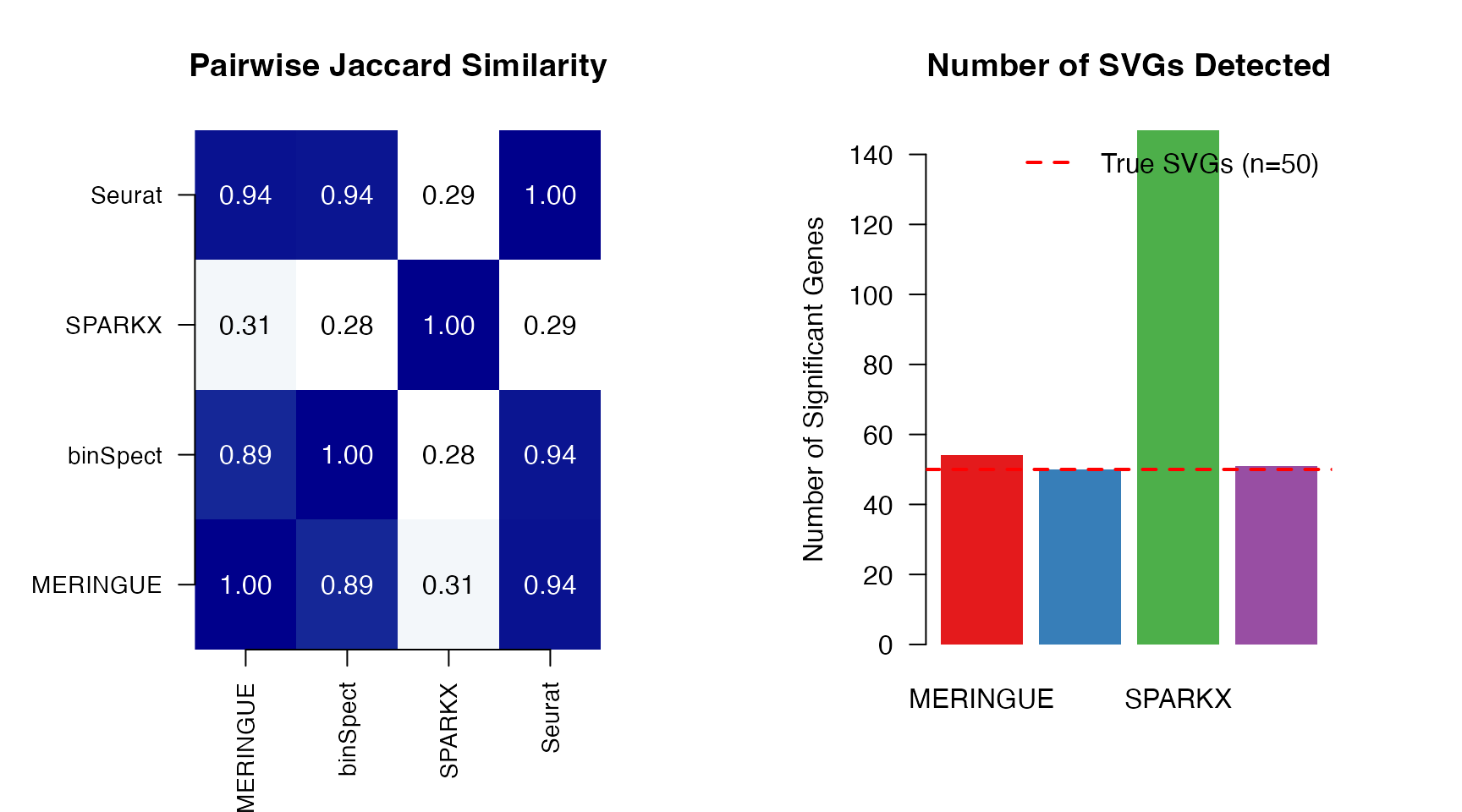
Advanced Analysis
Data Simulation for Custom Benchmarking
Generate custom simulated datasets with controlled parameters:
# Set seed for reproducibility
set.seed(2024)
# Generate custom dataset with specific parameters
sim_data <- simulate_spatial_data(
n_spots = 400, # Number of spatial locations
n_genes = 150, # Total genes
n_svg = 30, # Number of true SVGs
grid_type = "hexagonal", # Spatial arrangement
pattern_types = c("gradient", "hotspot", "periodic"), # Pattern types to include
mean_counts = 150, # Mean expression level
dispersion = 2.5 # Negative binomial dispersion
)
cat("Custom Simulation Summary:\n")
#> Custom Simulation Summary:
cat(" Spots:", ncol(sim_data$counts), "\n")
#> Spots: 400
cat(" Genes:", nrow(sim_data$counts), "\n")
#> Genes: 150
cat(" True SVGs:", sum(sim_data$gene_info$is_svg), "\n")
#> True SVGs: 30
cat("\nPattern distribution:\n")
#>
#> Pattern distribution:
print(table(sim_data$gene_info$pattern_type))
#>
#> gradient hotspot none periodic
#> 10 10 120 10Parallelization for Large Datasets
# Demonstrate parallel processing
# Note: mclapply doesn't work on Windows; falls back to sequential
n_cores <- 2 # Adjust based on your system
t_start <- Sys.time()
results_parallel <- CalSVG_MERINGUE(
expr_matrix = expr_log,
spatial_coords = coords,
n_threads = n_cores,
verbose = FALSE
)
t_end <- Sys.time()
cat(sprintf("Parallel execution with %d cores: %.2f seconds\n",
n_cores, as.numeric(t_end - t_start, units = "secs")))
#> Parallel execution with 2 cores: 0.03 secondsGene Filtering Strategies
# Pre-filter genes to improve signal-to-noise and reduce computation
# Strategy 1: Expression threshold
gene_means <- rowMeans(expr_log)
expr_high <- expr_log[gene_means > quantile(gene_means, 0.1), ]
cat("After expression filter:", nrow(expr_high), "genes\n")
#> After expression filter: 180 genes
# Strategy 2: Variance filter
gene_vars <- apply(expr_high, 1, var)
expr_filtered <- expr_high[gene_vars > quantile(gene_vars, 0.25), ]
cat("After variance filter:", nrow(expr_filtered), "genes\n")
#> After variance filter: 135 genes
# Strategy 3: Coefficient of variation
cv <- apply(expr_high, 1, sd) / (rowMeans(expr_high) + 0.1)
expr_cv <- expr_high[cv > quantile(cv, 0.25), ]
cat("After CV filter:", nrow(expr_cv), "genes\n")
#> After CV filter: 135 genesPractical Guidelines
Method Selection Framework
| Scenario | Recommended Method | Rationale |
|---|---|---|
| Exploratory analysis | binSpect or Seurat | Fast, good general performance |
| Manuscript/publication | Multiple methods + consensus | Robust, reproducible |
| Large datasets (>10k spots) | SPARK-X | Scalable, efficient |
| Effect size needed | nnSVG | Provides prop_sv metric |
| Multiple pattern types expected | SPARK-X | Multi-kernel detection |
| Seurat workflow integration | CalSVG_Seurat | Compatible output format |
Conclusion
The SVG package provides a comprehensive toolkit for
spatially variable gene detection in spatial transcriptomics data. Key
features include:
-
Unified Interface: Single function
(
CalSVG()) to access all methods - Rigorous Statistics: Proper null hypothesis testing and multiple testing correction
- Scalability: C++ acceleration and parallelization support
- Flexibility: Extensive parameter customization for diverse experimental designs
- Reproducibility: Consistent output format across all methods
For questions, bug reports, or feature requests, please visit: https://github.com/Zaoqu-Liu/SVG
Session Information
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] SVG_1.0.0
#>
#> loaded via a namespace (and not attached):
#> [1] cli_3.6.5 knitr_1.51 rlang_1.1.7
#> [4] xfun_0.56 otel_0.2.0 geometry_0.5.2
#> [7] textshaping_1.0.4 jsonlite_2.0.0 htmltools_0.5.9
#> [10] CompQuadForm_1.4.4 ragg_1.5.0 sass_0.4.10
#> [13] rmarkdown_2.30 abind_1.4-8 evaluate_1.0.5
#> [16] jquerylib_0.1.4 MASS_7.3-65 fastmap_1.2.0
#> [19] yaml_2.3.12 lifecycle_1.0.5 magic_1.6-1
#> [22] compiler_4.4.0 codetools_0.2-20 fs_1.6.6
#> [25] htmlwidgets_1.6.4 Rcpp_1.1.1 BiocParallel_1.40.2
#> [28] systemfonts_1.3.1 digest_0.6.39 R6_2.6.1
#> [31] RANN_2.6.2 parallel_4.4.0 bslib_0.9.0
#> [34] tools_4.4.0 pkgdown_2.1.3 cachem_1.1.0
#> [37] desc_1.4.3