SpaTalk for Different Spatial Transcriptomics Platforms
Zaoqu Liu
Maintainerliuzaoqu@163.com
2026-01-23
Source:vignettes/platforms.Rmd
platforms.RmdOverview
Spatial transcriptomics technologies can be broadly categorized into two types:
| Category | Resolution | Examples | SpaTalk Setting |
|---|---|---|---|
| Spot-based | Multi-cellular | 10x Visium, Slide-seq, ST | if_st_is_sc = FALSE |
| Single-cell | Cellular | STARmap, MERFISH, seqFISH+, Xenium | if_st_is_sc = TRUE |
This vignette provides platform-specific guidance for using SpaTalk.
Single-Cell Resolution Platforms
STARmap Example (Built-in Data)
STARmap provides single-cell resolution with targeted gene panels. Let’s demonstrate with the built-in STARmap data:
library(SpaTalk)
# Load built-in STARmap demo data
load(system.file("extdata", "starmap_data.rda", package = "SpaTalk"))
load(system.file("extdata", "starmap_meta.rda", package = "SpaTalk"))
# Check data dimensions
cat("Genes:", nrow(starmap_data), "\n")
#> Genes: 996
cat("Cells:", ncol(starmap_data), "\n")
#> Cells: 930
cat("Cell types:", length(unique(starmap_meta$celltype)), "\n")
#> Cell types: 14
# Create SpaTalk object - NO deconvolution needed
st_meta <- data.frame(
cell = starmap_meta$cell,
x = starmap_meta$x,
y = starmap_meta$y
)
obj <- createSpaTalk(
st_data = starmap_data,
st_meta = st_meta,
species = "Mouse",
if_st_is_sc = TRUE, # Single-cell resolution
spot_max_cell = 1, # One cell per "spot"
celltype = starmap_meta$celltype # Direct annotation
)
obj
#> An object of class SpaTalk
#> 996 genes across 930 single-cells (0 lrpair)
plot_st_celltype_all(obj, size = 1.2)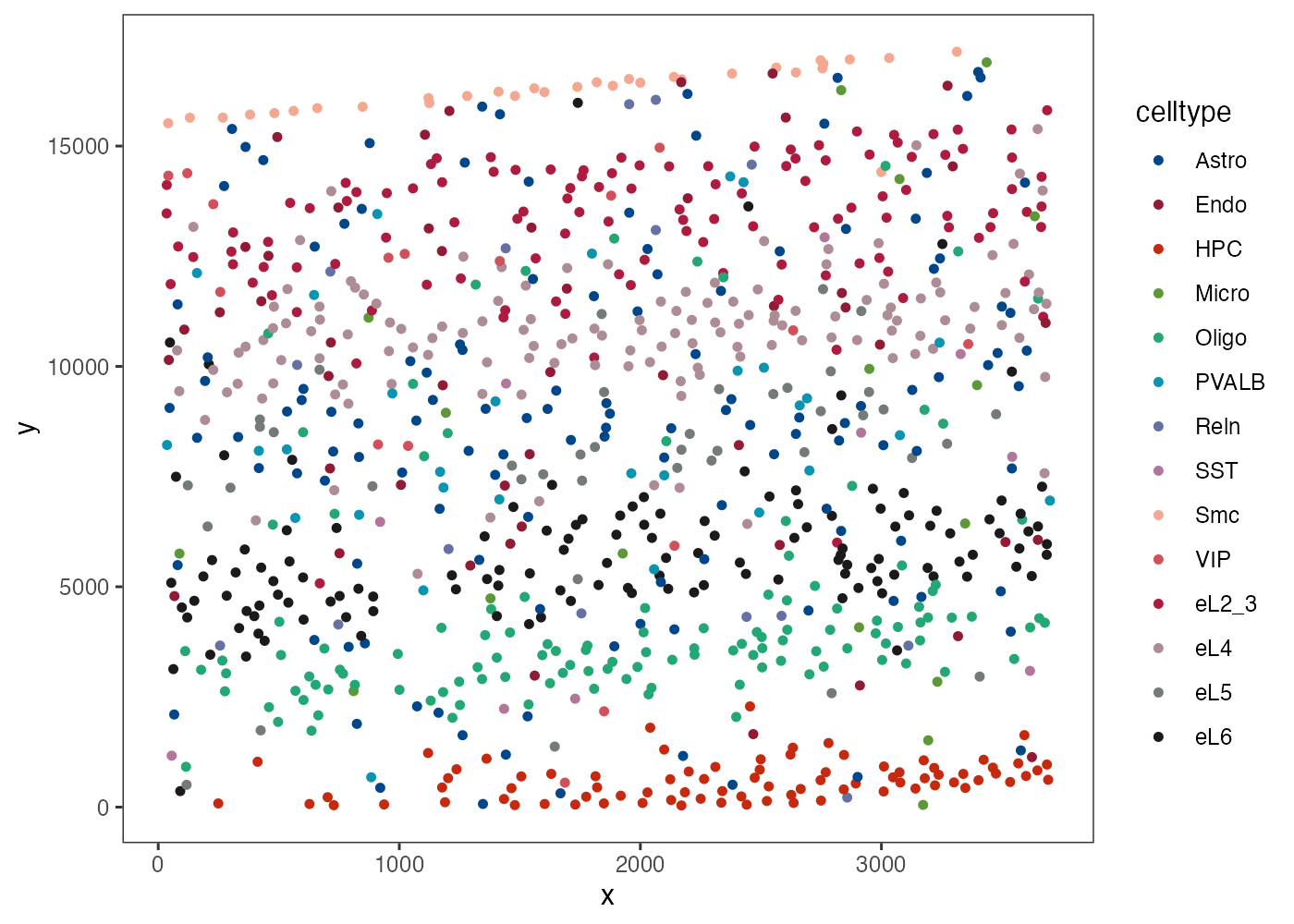
STARmap single-cell spatial distribution
Other Single-Cell Platforms
For MERFISH, Xenium, seqFISH+, etc., use the same workflow:
# Generic single-cell resolution workflow
obj <- createSpaTalk(
st_data = your_counts, # Gene x Cell matrix
st_meta = your_coords, # data.frame with cell, x, y
species = "Human", # or "Mouse"
if_st_is_sc = TRUE, # Single-cell resolution
spot_max_cell = 1,
celltype = your_annotations # Cell type labels
)
# No deconvolution needed - proceed directly to CCI
data(lrpairs)
data(pathways)
obj <- find_lr_path(obj, lrpairs, pathways)
obj <- dec_cci_all(obj)Spot-Based Platforms
10x Visium Workflow
Visium captures ~1-10 cells per 55μm diameter spot with whole-transcriptome coverage.
library(SpaTalk)
library(Seurat)
# Load Visium data using Seurat
visium <- Load10X_Spatial(
data.dir = "path/to/spaceranger/output/",
filename = "filtered_feature_bc_matrix.h5"
)
# Quality control
visium <- subset(visium,
nFeature_Spatial > 200 &
nFeature_Spatial < 10000 &
percent.mt < 20
)
# Extract for SpaTalk
st_data <- GetAssayData(visium, slot = "counts")
coords <- GetTissueCoordinates(visium)
st_meta <- data.frame(
spot = rownames(coords),
x = coords$imagerow,
y = coords$imagecol
)
# Create SpaTalk object (spot-based)
obj <- createSpaTalk(
st_data = st_data,
st_meta = st_meta,
species = "Human",
if_st_is_sc = FALSE, # Spot-based
spot_max_cell = 8 # Visium: ~1-10 cells/spot
)
# Deconvolution with scRNA-seq reference (REQUIRED for spot-based)
obj <- dec_celltype(
object = obj,
sc_data = sc_reference,
sc_celltype = sc_annotations
)Slide-seq / Slide-seqV2
Slide-seq uses 10μm beads, typically capturing 1-10 cells per bead.
# Slide-seq workflow
obj <- createSpaTalk(
st_data = slideseq_counts,
st_meta = data.frame(
spot = colnames(slideseq_counts),
x = bead_coordinates$x,
y = bead_coordinates$y
),
species = "Mouse",
if_st_is_sc = FALSE,
spot_max_cell = 5 # Slide-seq: smaller spots
)
# Deconvolution required
obj <- dec_celltype(obj, sc_data, sc_celltype)Workflow Comparison
Single-cell Workflow (STARmap, MERFISH, Xenium)
# Complete single-cell workflow demonstration
library(SpaTalk)
# 1. Load data
load(system.file("extdata", "starmap_data.rda", package = "SpaTalk"))
load(system.file("extdata", "starmap_meta.rda", package = "SpaTalk"))
# 2. Create object with cell types
st_meta <- data.frame(
cell = starmap_meta$cell,
x = starmap_meta$x,
y = starmap_meta$y
)
obj <- createSpaTalk(
st_data = starmap_data,
st_meta = st_meta,
species = "Mouse",
if_st_is_sc = TRUE,
spot_max_cell = 1,
celltype = starmap_meta$celltype
)
# 3. NO deconvolution needed
# 4. Filter LR paths
data(lrpairs)
data(pathways)
obj <- find_lr_path(obj, lrpairs, pathways, if_doParallel = FALSE)
#> Checking input data
#> Begin to filter lrpairs and pathways
#> ***Done***
#>
# 5. Infer CCIs
obj <- dec_cci(obj, "eL6", "PVALB", if_doParallel = FALSE)
#> Begin to find LR pairs
#>
cat("Single-cell workflow completed!\n")
#> Single-cell workflow completed!
cat("LR pairs found:", nrow(obj@lrpair), "\n")
#> LR pairs found: 1Spot-based Workflow (Visium, Slide-seq)
# Spot-based workflow (requires scRNA-seq reference)
# 1. Create object (spot-based)
obj <- createSpaTalk(st_data, st_meta, "Human",
if_st_is_sc = FALSE, spot_max_cell = 10)
# 2. Deconvolution (REQUIRED)
obj <- dec_celltype(obj, sc_data, sc_celltype)
# 3. Filter LR paths
obj <- find_lr_path(obj, lrpairs, pathways)
# 4. Infer CCIs
obj <- dec_cci_all(obj)Parameter Guidelines by Platform
| Platform | spot_max_cell |
Deconvolution | Notes |
|---|---|---|---|
| 10x Visium | 5-10 | Required | 55μm spots |
| Slide-seq | 3-8 | Required | 10μm beads |
| Slide-seqV2 | 3-8 | Required | Improved capture |
| Original ST | 15-30 | Required | 100μm spots |
| STARmap | 1 | Not needed | Single-cell |
| MERFISH | 1 | Not needed | Single-cell |
| Xenium | 1 | Not needed | Single-cell |
| seqFISH+ | 1 | Not needed | Single-cell |
| CosMx | 1 | Not needed | Single-cell |
Best Practices
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] parallel stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] SpaTalk_2.0.0 doParallel_1.0.17 iterators_1.0.14 foreach_1.5.2
#> [5] ggalluvial_0.12.5 ggplot2_4.0.1
#>
#> loaded via a namespace (and not attached):
#> [1] RColorBrewer_1.1-3 jsonlite_2.0.0 magrittr_2.0.4
#> [4] spatstat.utils_3.2-1 farver_2.1.2 rmarkdown_2.30
#> [7] fs_1.6.6 ragg_1.5.0 vctrs_0.7.0
#> [10] ROCR_1.0-11 spatstat.explore_3.6-0 rstatix_0.7.3
#> [13] htmltools_0.5.9 progress_1.2.3 broom_1.0.11
#> [16] Formula_1.2-5 sass_0.4.10 sctransform_0.4.3
#> [19] parallelly_1.46.1 KernSmooth_2.23-26 bslib_0.9.0
#> [22] htmlwidgets_1.6.4 desc_1.4.3 ica_1.0-3
#> [25] plyr_1.8.9 plotly_4.11.0 zoo_1.8-15
#> [28] cachem_1.1.0 igraph_2.2.1 mime_0.13
#> [31] lifecycle_1.0.5 pkgconfig_2.0.3 Matrix_1.7-4
#> [34] R6_2.6.1 fastmap_1.2.0 fitdistrplus_1.2-4
#> [37] future_1.69.0 shiny_1.12.1 digest_0.6.39
#> [40] patchwork_1.3.2 Seurat_4.4.0 tensor_1.5.1
#> [43] irlba_2.3.5.1 textshaping_1.0.4 ggpubr_0.6.2
#> [46] labeling_0.4.3 progressr_0.18.0 spatstat.sparse_3.1-0
#> [49] httr_1.4.7 polyclip_1.10-7 abind_1.4-8
#> [52] compiler_4.4.0 withr_3.0.2 backports_1.5.0
#> [55] S7_0.2.1 carData_3.0-5 ggforce_0.5.0
#> [58] ggsignif_0.6.4 MASS_7.3-65 rappdirs_0.3.4
#> [61] ggsci_4.2.0 tools_4.4.0 lmtest_0.9-40
#> [64] otel_0.2.0 scatterpie_0.2.6 httpuv_1.6.16
#> [67] future.apply_1.20.1 goftest_1.2-3 glue_1.8.0
#> [70] nlme_3.1-168 promises_1.5.0 grid_4.4.0
#> [73] Rtsne_0.17 cluster_2.1.8.1 reshape2_1.4.5
#> [76] generics_0.1.4 gtable_0.3.6 spatstat.data_3.1-9
#> [79] tzdb_0.5.0 tidyr_1.3.2 data.table_1.18.0
#> [82] hms_1.1.4 car_3.1-3 sp_2.2-0
#> [85] spatstat.geom_3.6-1 RcppAnnoy_0.0.23 ggrepel_0.9.6
#> [88] RANN_2.6.2 pillar_1.11.1 stringr_1.6.0
#> [91] yulab.utils_0.2.3 ggExtra_0.11.0 later_1.4.5
#> [94] splines_4.4.0 tweenr_2.0.3 dplyr_1.1.4
#> [97] lattice_0.22-7 survival_3.8-3 deldir_2.0-4
#> [100] tidyselect_1.2.1 miniUI_0.1.2 pbapply_1.7-4
#> [103] knitr_1.51 gridExtra_2.3 scattermore_1.2
#> [106] xfun_0.56 matrixStats_1.5.0 pheatmap_1.0.13
#> [109] stringi_1.8.7 ggfun_0.2.0 lazyeval_0.2.2
#> [112] yaml_2.3.12 evaluate_1.0.5 codetools_0.2-20
#> [115] tibble_3.3.1 cli_3.6.5 uwot_0.2.4
#> [118] xtable_1.8-4 reticulate_1.44.1 systemfonts_1.3.1
#> [121] jquerylib_0.1.4 dichromat_2.0-0.1 Rcpp_1.1.1
#> [124] globals_0.18.0 spatstat.random_3.4-3 png_0.1-8
#> [127] spatstat.univar_3.1-6 readr_2.1.6 pkgdown_2.1.3
#> [130] NNLM_0.4.4 prettyunits_1.2.0 listenv_0.10.0
#> [133] viridisLite_0.4.2 scales_1.4.0 ggridges_0.5.7
#> [136] SeuratObject_4.1.4 leiden_0.4.3.1 purrr_1.2.1
#> [139] crayon_1.5.3 rlang_1.1.7 cowplot_1.2.0