Overview
This guide demonstrates the essential COMMOTR workflow in 5 simple steps: 1. Load ligand-receptor database 2. Infer spatial communication 3. Compute communication direction 4. Analyze cluster communication 5. Visualize results
Installation
# From R-universe (recommended)
install.packages("COMMOTR", repos = "https://zaoqu-liu.r-universe.dev")
# From GitHub
remotes::install_github("Zaoqu-Liu/COMMOTR")Create Demo Data
For this tutorial, we create a simulated spatial dataset with known communication patterns:
set.seed(42)
# Simulate 100 cells in two spatial clusters
n_cells <- 100
n_genes <- 50
# Create expression matrix
expr <- matrix(rpois(n_cells * n_genes, lambda = 5),
nrow = n_genes, ncol = n_cells)
# Gene names including ligand-receptor pairs
gene_names <- c("Tgfb1", "Tgfbr1", "Tgfbr2", # TGFb pathway
"Wnt5a", "Fzd1", "Lrp5", # Wnt pathway
"Fgf2", "Fgfr1", # FGF pathway
paste0("Gene", 1:(n_genes - 8)))
rownames(expr) <- gene_names
colnames(expr) <- paste0("Cell", 1:n_cells)
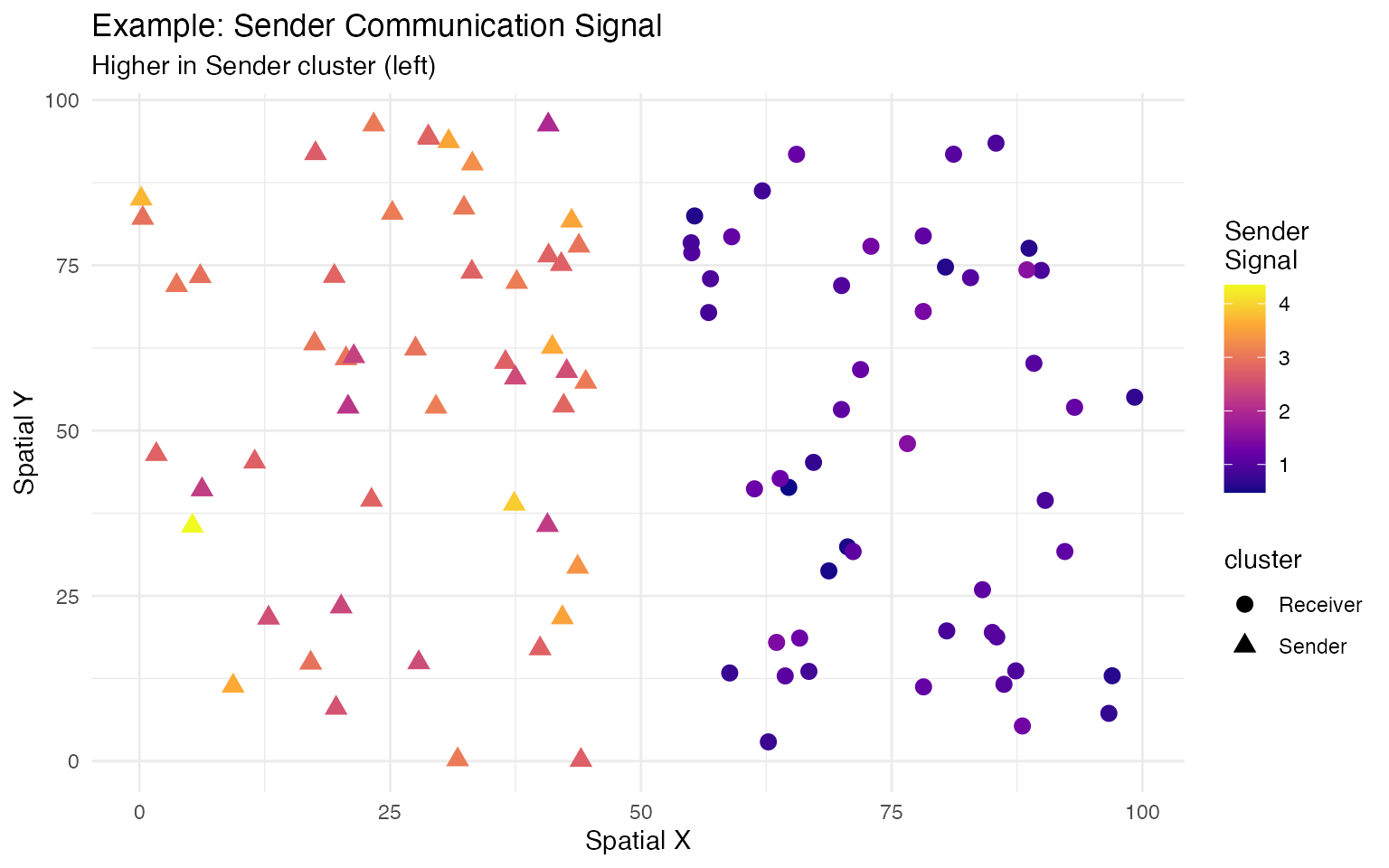
# Create spatial structure: left cluster (ligand-high), right cluster (receptor-high)
coords <- matrix(c(
runif(50, 0, 45), runif(50, 55, 100), # x coordinates
runif(100, 0, 100) # y coordinates
), ncol = 2)
rownames(coords) <- colnames(expr)
colnames(coords) <- c("x", "y")
# Add differential expression
expr["Tgfb1", 1:50] <- expr["Tgfb1", 1:50] + 15
expr["Tgfbr1", 51:100] <- expr["Tgfbr1", 51:100] + 15
expr["Tgfbr2", 51:100] <- expr["Tgfbr2", 51:100] + 15
# Create Seurat object
seurat_obj <- CreateSeuratObject(counts = expr, assay = "RNA")
seurat_obj <- SetAssayData(seurat_obj, layer = "data",
new.data = log1p(as(expr, "dgCMatrix")))
# Add spatial coordinates
colnames(coords) <- c("spatial_1", "spatial_2")
seurat_obj[["spatial"]] <- CreateDimReducObject(
embeddings = coords, key = "spatial_", assay = "RNA"
)
# Add cluster labels
seurat_obj$cluster <- factor(c(rep("Sender", 50), rep("Receiver", 50)))Step 1: Load LR Database
# Create custom LR database for demo
df_lr <- data.frame(
ligand = c("Tgfb1", "Wnt5a", "Fgf2"),
receptor = c("Tgfbr1_Tgfbr2", "Fzd1_Lrp5", "Fgfr1"),
pathway = c("TGFb", "WNT", "FGF"),
stringsAsFactors = FALSE
)
# In real analysis, load from built-in databases:
df_lr <- ligand_receptor_database("CellChat", "mouse", "Secreted Signaling")
df_lr <- filter_lr_database(df_lr, seurat_obj, min_cell_pct = 0.05)Step 2: Infer Spatial Communication
# Run spatial communication inference
seurat_obj <- spatial_communication(
seurat_obj,
df_ligrec = df_lr,
database_name = "demo",
spatial_coords = "spatial",
dis_thr = 40, # Distance threshold
cot_eps_p = 0.1, # Entropy regularization
cot_rho = 10, # Unbalanced penalty
verbose = TRUE
)
# Access results
results <- get_communication_results(seurat_obj, "demo")Step 3: Communication Direction
# Compute vector field for TGFb pathway
seurat_obj <- communication_direction(
seurat_obj,
database_name = "demo",
pathway_name = "TGFb",
spatial_coords = "spatial",
k = 5
)Step 4: Cluster Communication
# Analyze cluster-level communication with permutation test
seurat_obj <- cluster_communication(
seurat_obj,
database_name = "demo",
clustering = "cluster",
pathway_name = "TGFb",
n_permutations = 100
)
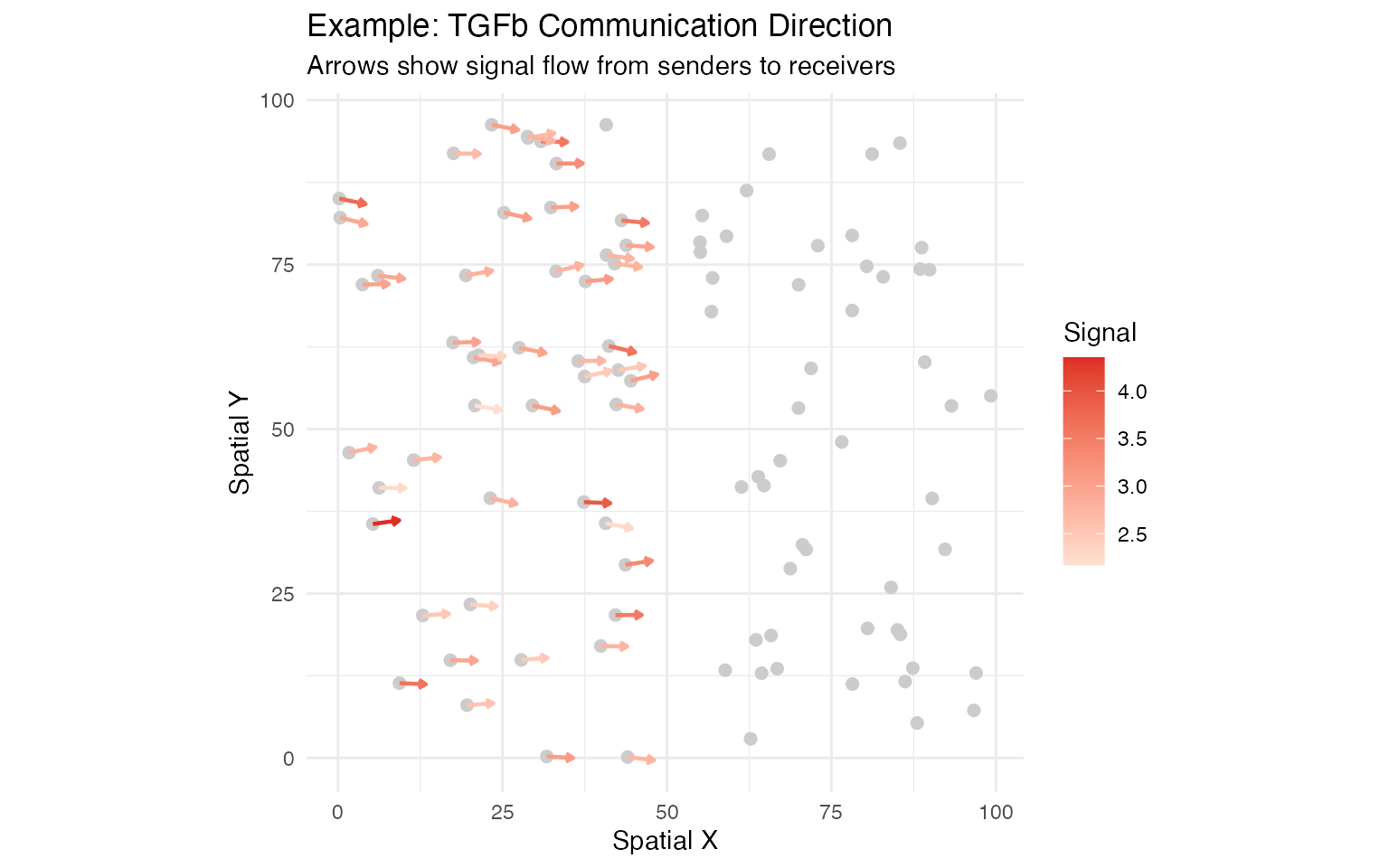
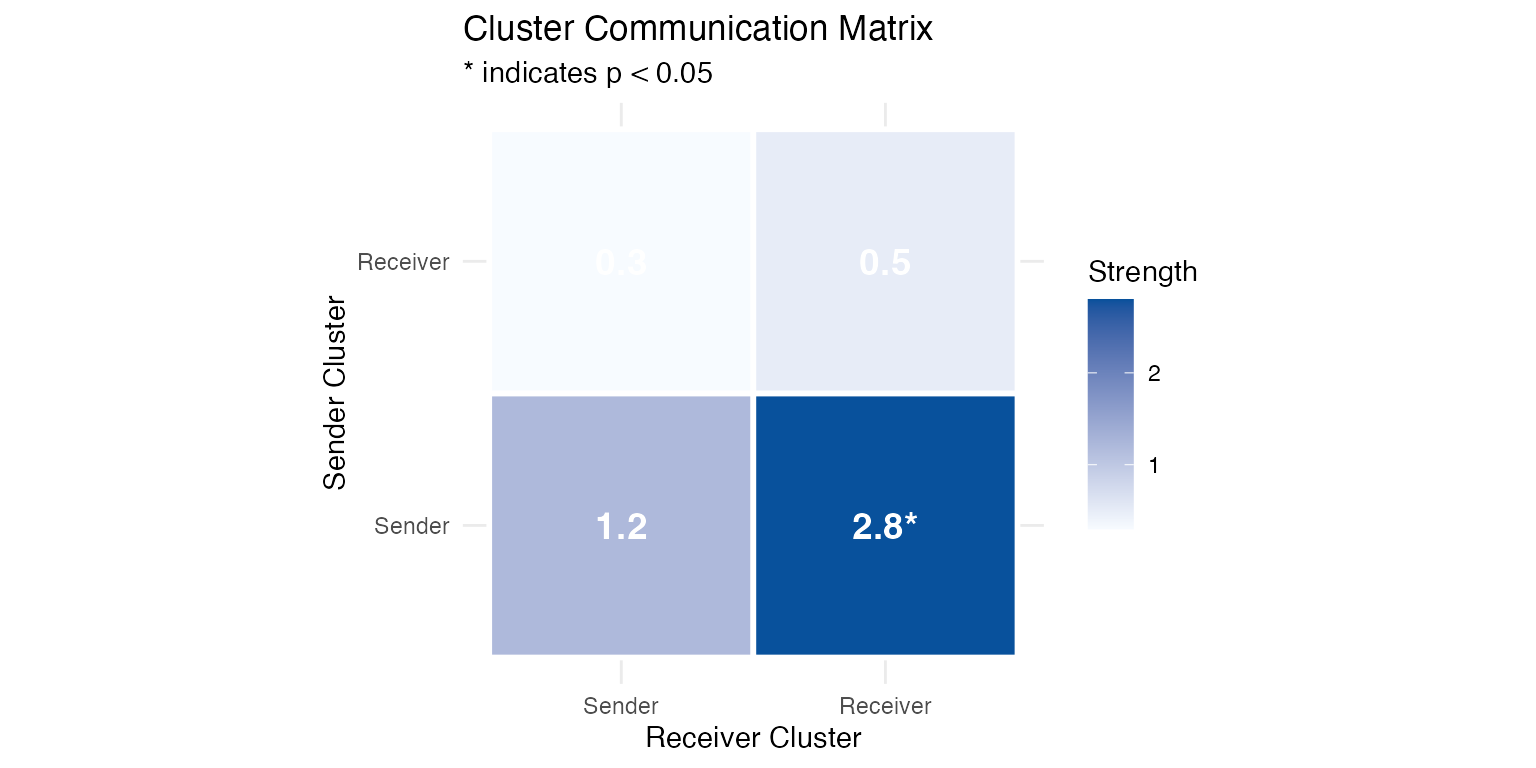
Summary
| Step | Function | Purpose |
|---|---|---|
| 1 | ligand_receptor_database() |
Load LR pairs |
| 2 | spatial_communication() |
Infer communication |
| 3 | communication_direction() |
Compute vector fields |
| 4 | cluster_communication() |
Cluster-level analysis |
| 5 | Visualization | Interpret results |
Next Steps
- Read the Algorithm Theory vignette for mathematical details
- Explore the Visualization Gallery for more plot types
- Check the Full Tutorial for advanced analysis
Developed by Zaoqu Liu | GitHub | liuzaoqu@163.com