Advanced Usage and Best Practices
Zaoqu Liu
2026-01-29
Source:vignettes/advanced-usage.Rmd
advanced-usage.RmdIntroduction
This vignette covers advanced usage patterns, parameter tuning, and best practices for using SCORPION effectively.
Parameter Tuning
Gamma Value (Metacell Aggregation)
The gammaValue parameter controls how many cells are
aggregated into each metacell:
# Test different gamma values
gammas <- c(5, 10, 20)
results_gamma <- list()
for(g in gammas) {
set.seed(123)
results_gamma[[as.character(g)]] <- scorpion(
tfMotifs = scorpionTest$tf,
gexMatrix = scorpionTest$gex,
ppiNet = scorpionTest$ppi,
gammaValue = g,
alphaValue = 0.5,
hammingValue = 0.01,
showProgress = FALSE
)
}
# Compare network statistics
cat("Effect of gamma on network properties:\n")
#> Effect of gamma on network properties:
for(g in gammas) {
r <- results_gamma[[as.character(g)]]
cat(sprintf("gamma=%d: mean=%.3f, sd=%.3f\n",
g, mean(r$regNet), sd(r$regNet)))
}
#> gamma=5: mean=-0.004, sd=0.981
#> gamma=10: mean=-0.004, sd=0.977
#> gamma=20: mean=-0.006, sd=0.996Guidelines: - γ = 5-10: For small
datasets (< 1000 cells) - γ = 10-20: For medium
datasets (1000-10000 cells)
- γ = 20-50: For large datasets (> 10000 cells)
Alpha Value (Learning Rate)
The alphaValue controls how quickly networks are
updated:
# Test different alpha values
alphas <- c(0.05, 0.1, 0.2)
results_alpha <- list()
for(a in alphas) {
set.seed(123)
results_alpha[[as.character(a)]] <- scorpion(
tfMotifs = scorpionTest$tf,
gexMatrix = scorpionTest$gex,
ppiNet = scorpionTest$ppi,
gammaValue = 10,
alphaValue = a,
hammingValue = 0.001,
showProgress = FALSE
)
}
# Compare convergence behavior
cat("Effect of alpha on convergence:\n")
#> Effect of alpha on convergence:
for(a in alphas) {
r <- results_alpha[[as.character(a)]]
cat(sprintf("alpha=%.2f: edges=%d\n", a, r$numEdges))
}
#> alpha=0.05: edges=167562
#> alpha=0.10: edges=167562
#> alpha=0.20: edges=167562Guidelines: - α = 0.05-0.1: Conservative, slower convergence but more stable - α = 0.1-0.2: Default, good balance - α > 0.2: Aggressive, may be unstable
Hamming Threshold
The hammingValue determines convergence sensitivity:
# Test different hamming thresholds
hammings <- c(0.01, 0.001, 0.0001)
set.seed(123)
result_h1 <- scorpion(
tfMotifs = scorpionTest$tf,
gexMatrix = scorpionTest$gex,
ppiNet = scorpionTest$ppi,
gammaValue = 10,
alphaValue = 0.1,
hammingValue = 0.01,
showProgress = FALSE
)
set.seed(123)
result_h2 <- scorpion(
tfMotifs = scorpionTest$tf,
gexMatrix = scorpionTest$gex,
ppiNet = scorpionTest$ppi,
gammaValue = 10,
alphaValue = 0.1,
hammingValue = 0.001,
showProgress = FALSE
)
# Compare results
cor_reg <- cor(as.vector(result_h1$regNet), as.vector(result_h2$regNet))
cat("Correlation between hamming=0.01 and 0.001:", round(cor_reg, 4), "\n")
#> Correlation between hamming=0.01 and 0.001: 1Association Methods
SCORPION supports three methods for computing gene co-expression:
Comparison
par(mfrow = c(1, 3), mar = c(4, 4, 3, 1))
hist(as.vector(result_pearson$regNet), breaks = 50,
main = "Pearson", xlab = "Edge Weight", col = "steelblue")
hist(as.vector(result_spearman$regNet), breaks = 50,
main = "Spearman", xlab = "Edge Weight", col = "coral")
hist(as.vector(result_pcnet$regNet), breaks = 50,
main = "pcNet", xlab = "Edge Weight", col = "forestgreen")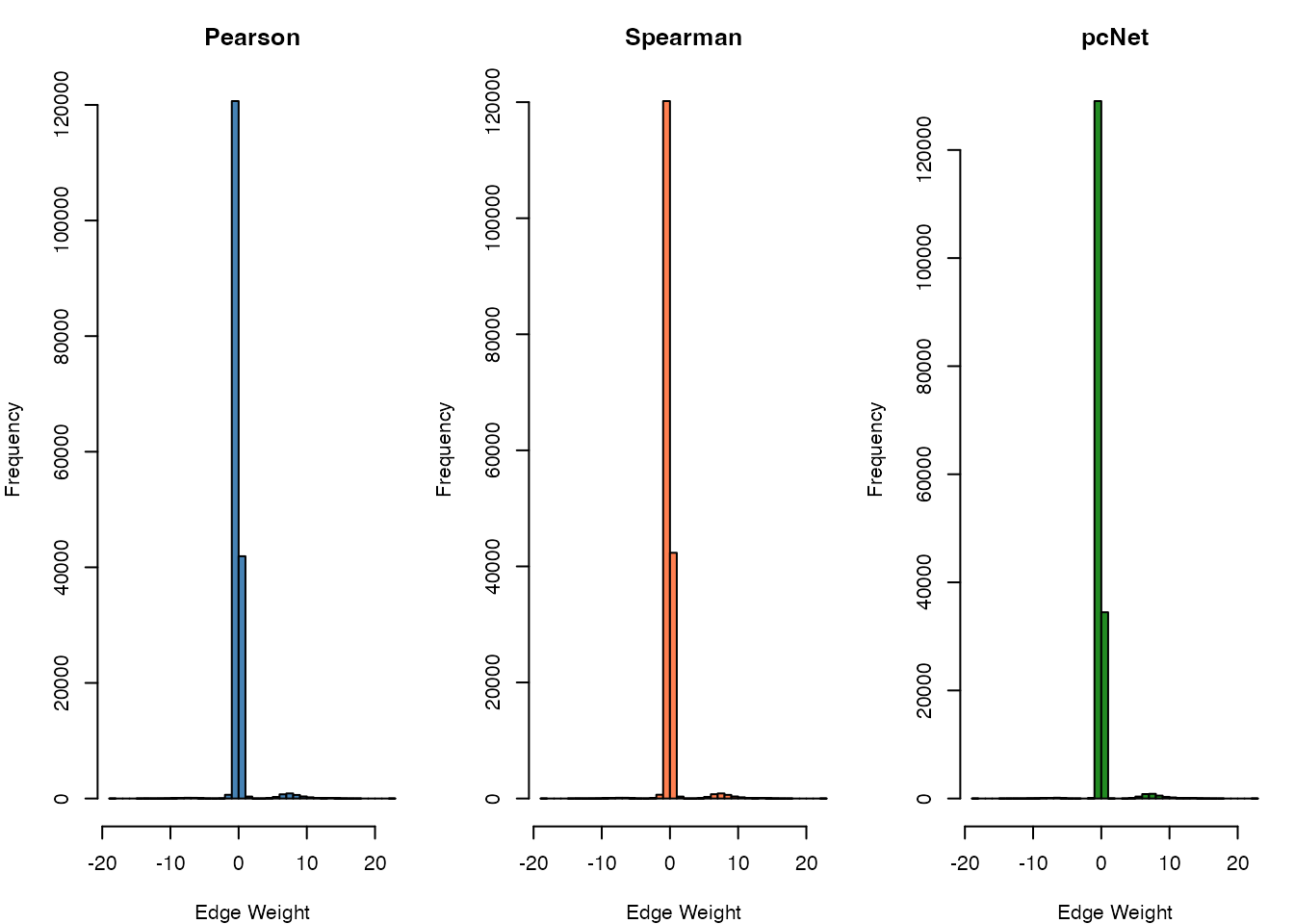
Comparison of association methods
Working with Seurat Objects
If you have a Seurat object, extract the expression matrix:
# Example with Seurat object
library(Seurat)
# Extract expression matrix from Seurat v4
gex_matrix <- GetAssayData(seurat_obj, slot = "counts")
# Or from Seurat v5
gex_matrix <- seurat_obj[["RNA"]]$counts
# Run SCORPION
result <- scorpion(
tfMotifs = tf_data,
gexMatrix = gex_matrix,
ppiNet = ppi_data
)Filtering and Preprocessing
Filter Low-Expression Genes
# Filter genes before running SCORPION
set.seed(123)
result_filtered <- scorpion(
tfMotifs = scorpionTest$tf,
gexMatrix = scorpionTest$gex,
ppiNet = scorpionTest$ppi,
filterExpr = TRUE, # Remove zero-expression genes
showProgress = FALSE
)
cat("Genes after filtering:", result_filtered$numGenes, "\n")
#> Genes after filtering: 214Custom Gene Filtering
# Manual filtering
gex <- scorpionTest$gex
# Keep genes expressed in at least 5% of cells
min_cells <- ncol(gex) * 0.05
gex_filtered <- gex[Matrix::rowSums(gex > 0) >= min_cells, ]
cat("Original genes:", nrow(scorpionTest$gex), "\n")
#> Original genes: 230
cat("Filtered genes:", nrow(gex_filtered), "\n")
#> Filtered genes: 225
# Run with filtered data
set.seed(123)
result_custom <- scorpion(
tfMotifs = scorpionTest$tf,
gexMatrix = gex_filtered,
ppiNet = scorpionTest$ppi,
showProgress = FALSE
)Parallel Processing
SCORPION supports parallel computation for the pcNet method:
# Use multiple cores
result_parallel <- scorpion(
tfMotifs = tf_data,
gexMatrix = gex_data,
ppiNet = ppi_data,
assocMethod = "pcNet",
nCores = 4 # Use 4 cores
)Output Analysis
Network Comparison
Compare networks across conditions:
# Simulate two conditions
set.seed(123)
result1 <- scorpion(
tfMotifs = scorpionTest$tf,
gexMatrix = scorpionTest$gex,
ppiNet = scorpionTest$ppi,
alphaValue = 0.1,
showProgress = FALSE
)
set.seed(456)
result2 <- scorpion(
tfMotifs = scorpionTest$tf,
gexMatrix = scorpionTest$gex,
ppiNet = scorpionTest$ppi,
alphaValue = 0.1,
showProgress = FALSE
)
# Compare regulatory networks
plot(as.vector(result1$regNet), as.vector(result2$regNet),
pch = ".", col = adjustcolor("steelblue", 0.3),
xlab = "Condition 1", ylab = "Condition 2",
main = "Regulatory Network Comparison")
abline(0, 1, col = "red", lty = 2)
# Correlation
r <- cor(as.vector(result1$regNet), as.vector(result2$regNet))
legend("bottomright", paste("r =", round(r, 3)), bty = "n")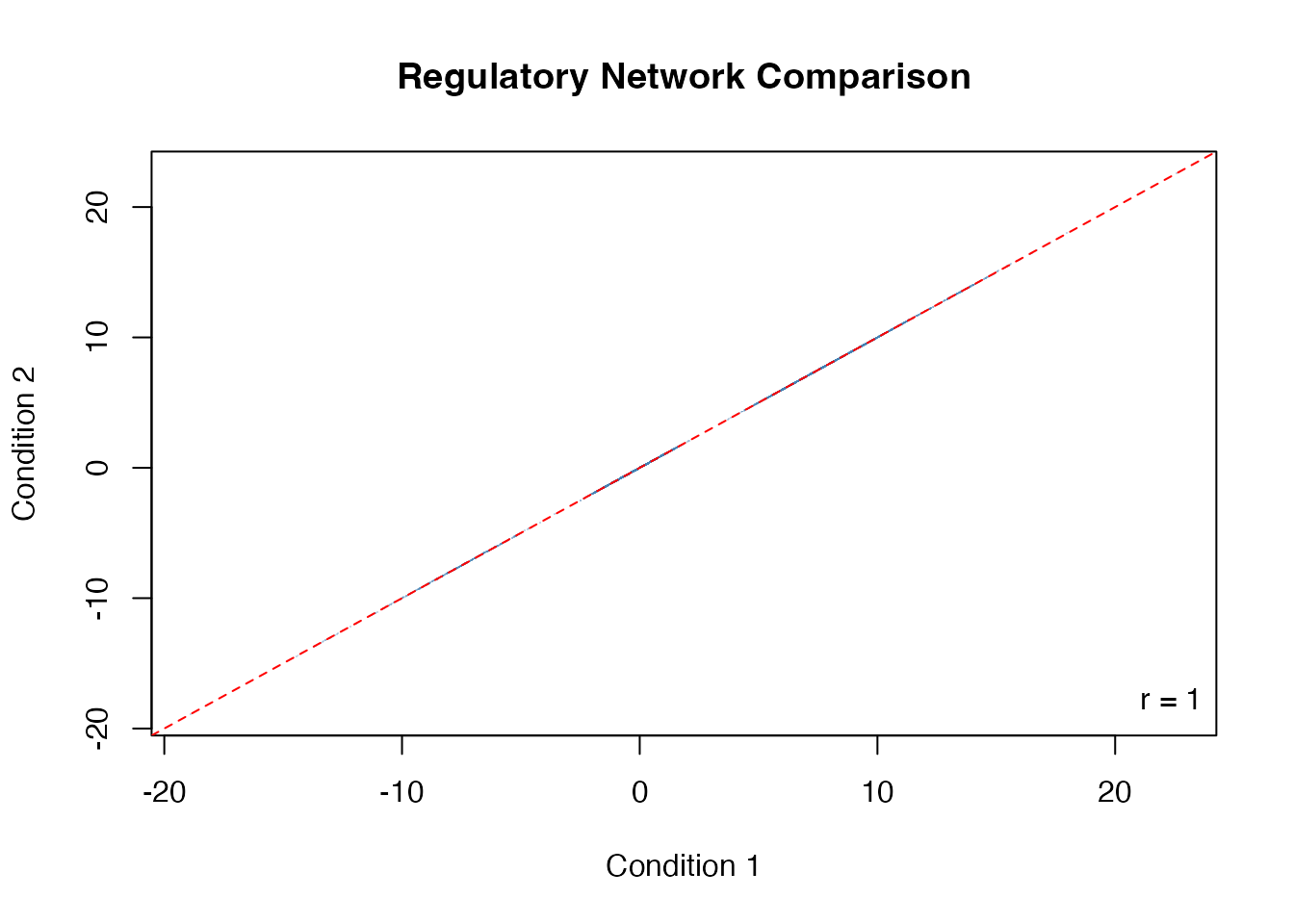
Network comparison across conditions
Differential Network Analysis
# Compute differential edges
diff_net <- result1$regNet - result2$regNet
# Find significantly different edges
threshold <- 2 * sd(diff_net)
diff_edges <- which(abs(diff_net) > threshold, arr.ind = TRUE)
if(nrow(diff_edges) > 0) {
diff_df <- data.frame(
TF = rownames(diff_net)[diff_edges[,1]],
Gene = colnames(diff_net)[diff_edges[,2]],
Difference = diff_net[diff_edges]
)
diff_df <- diff_df[order(-abs(diff_df$Difference)), ]
cat("Top differential edges:\n")
head(diff_df, 10)
}Best Practices
1. Data Quality
- Remove low-quality cells before running SCORPION
- Filter genes with very low expression
- Consider batch effects if analyzing multiple samples
2. Prior Network Selection
- Use species-appropriate TF-target databases
- Higher-confidence PPI scores improve results
- Consider tissue-specific priors when available
Session Information
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] Matrix_1.7-4 SCORPION_1.2.1
#>
#> loaded via a namespace (and not attached):
#> [1] cli_3.6.5 knitr_1.51 rlang_1.1.7 xfun_0.56
#> [5] otel_0.2.0 textshaping_1.0.4 jsonlite_2.0.0 htmltools_0.5.9
#> [9] ragg_1.5.0 sass_0.4.10 rmarkdown_2.30 grid_4.4.0
#> [13] evaluate_1.0.5 jquerylib_0.1.4 fastmap_1.2.0 yaml_2.3.12
#> [17] lifecycle_1.0.5 compiler_4.4.0 igraph_2.2.1 irlba_2.3.5.1
#> [21] fs_1.6.6 pkgconfig_2.0.3 htmlwidgets_1.6.4 pbapply_1.7-4
#> [25] systemfonts_1.3.1 lattice_0.22-7 digest_0.6.39 R6_2.6.1
#> [29] RANN_2.6.2 parallel_4.4.0 magrittr_2.0.4 bslib_0.9.0
#> [33] tools_4.4.0 pkgdown_2.2.0 cachem_1.1.0 desc_1.4.3