Documentation: https://zaoqu-liu.github.io/SCORPION/
What’s New 🎉
v1.2.2 (January 2026)
- 🚀 RcppArmadillo acceleration: C++ implementation of core algorithms with BLAS optimization (~2.5x faster)
- 📚 Documentation website: Full pkgdown site with tutorials at https://zaoqu-liu.github.io/SCORPION/
- 📖 Vignettes: 4 comprehensive guides (Quick Start, Algorithm, Advanced Usage, Visualization)
- ⚡ Performance: Vectorized operations + optimized BLAS for matrix computation
- 🛡️ Robustness: Enhanced numerical stability in Tanimoto similarity
- 🌐 R-universe: Easy installation via
install.packages("SCORPION", repos = "https://zaoqu-liu.r-universe.dev")
Overview
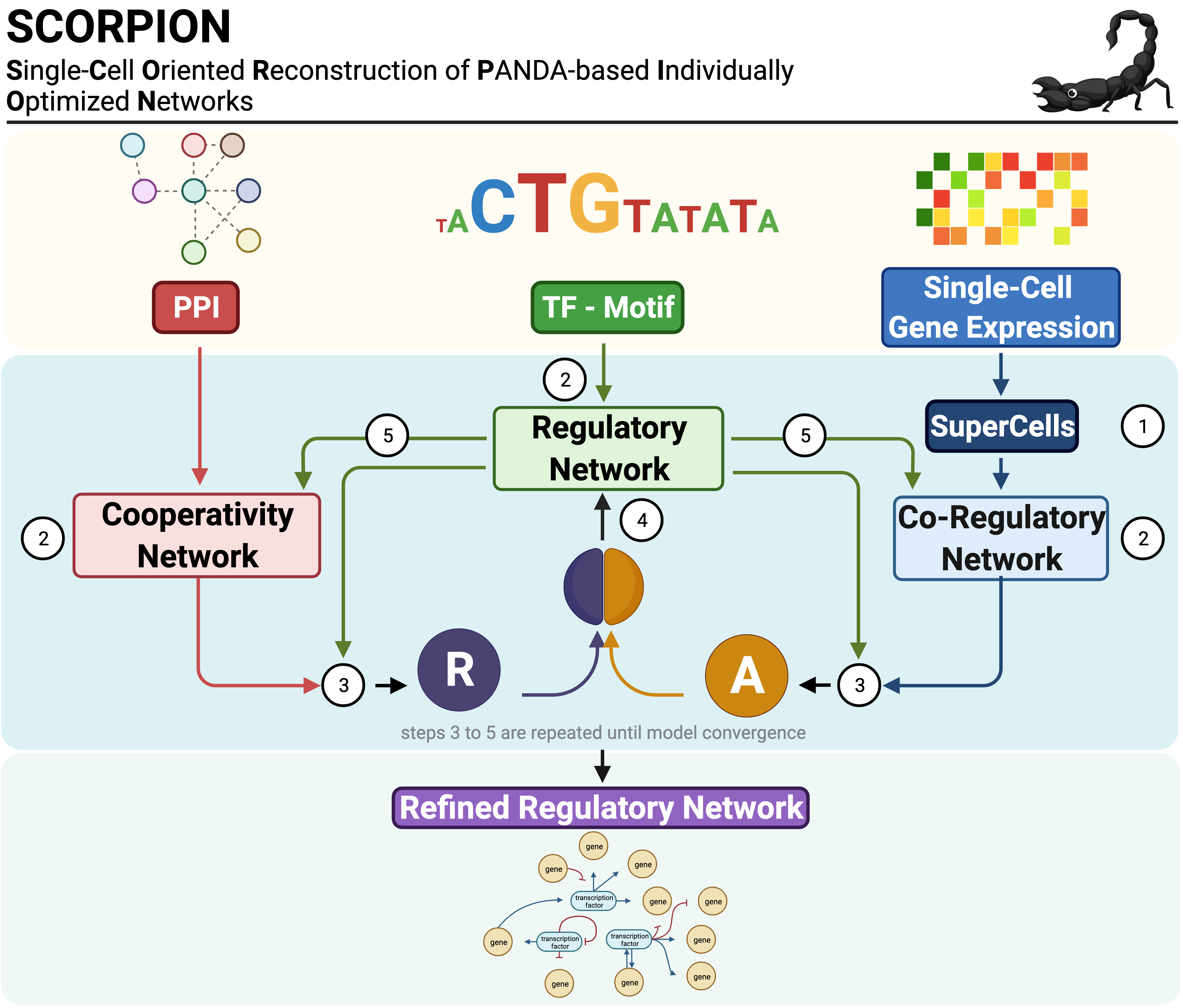
SCORPION (Single-Cell Oriented Reconstruction of PANDA Individually Optimized Gene Regulatory Networks) is a computational framework for inferring cell-type-specific gene regulatory networks (GRNs) from single-cell RNA sequencing (scRNA-seq) data.
Motivation
Single-cell transcriptomics has revolutionized our understanding of cellular heterogeneity, yet the inherent sparsity and technical noise in scRNA-seq data pose significant challenges for accurate gene regulatory network inference. SCORPION addresses these limitations through a metacell-based coarse-graining strategy that preserves biological signal while reducing technical artifacts.
Method
SCORPION integrates three complementary data modalities to reconstruct transcriptome-wide GRNs:
- Transcription factor (TF) binding motifs - sequence-based prior knowledge of potential regulatory relationships
- Protein-protein interactions (PPI) - physical interactions between transcription factors
- Gene expression profiles - cell-type-specific transcriptional states
The algorithm employs the PANDA (Passing Attributes between Networks for Data Assimilation) message-passing framework, which iteratively refines network edge weights through Tanimoto similarity-based updates until convergence.
Key Features
- Metacell aggregation: Reduces data sparsity by grouping transcriptionally similar cells using graph-based clustering in PCA space
- Multi-modal integration: Combines transcriptomic data with prior biological knowledge (TF motifs, PPI networks)
- Comparable networks: Generates fully-connected, weighted, and directed GRNs suitable for population-level comparative analyses
- Scalable: Efficient implementation supporting both CPU and GPU computation
Installation
From R-universe (Recommended)
install.packages("SCORPION", repos = "https://zaoqu-liu.r-universe.dev")From CRAN
install.packages("SCORPION")From GitHub (Development Version)
if (!requireNamespace("remotes", quietly = TRUE))
install.packages("remotes")
remotes::install_github("Zaoqu-Liu/SCORPION")Quick Start
library(SCORPION)
# Load example data
data(scorpionTest)
# Infer gene regulatory network
grn <- scorpion(
tfMotifs = scorpionTest$tf, # TF-target motif prior
gexMatrix = scorpionTest$gex, # Gene expression matrix (genes × cells)
ppiNet = scorpionTest$ppi, # Protein-protein interactions
alphaValue = 0.1 # Learning rate (default)
)Input Data Format
| Parameter | Description | Format |
|---|---|---|
tfMotifs |
TF-target regulatory prior | data.frame with columns: TF, Target, Score |
gexMatrix |
Gene expression matrix | matrix/dgCMatrix (genes × cells) |
ppiNet |
Protein-protein interactions | data.frame with columns: TF1, TF2, Score |
Output
SCORPION returns a list containing:
| Component | Description | Dimensions |
|---|---|---|
regNet |
TF-gene regulatory network | TFs × Genes |
coregNet |
Gene co-regulatory network | Genes × Genes |
coopNet |
TF cooperative network | TFs × TFs |
Edge weights represent Z-scores indicating the strength of regulatory relationships.
Example Workflow
# Load package and data
library(SCORPION)
data(scorpionTest)
# Run SCORPION
result <- scorpion(
tfMotifs = scorpionTest$tf,
gexMatrix = scorpionTest$gex,
ppiNet = scorpionTest$ppi,
gammaValue = 10, # Metacell graining level
nPC = 25, # Principal components for clustering
alphaValue = 0.1, # Learning rate
hammingValue = 0.001 # Convergence threshold
)
# Examine output
dim(result$regNet) # Regulatory network dimensions
# [1] 783 214
# Extract top regulatory edges
regNet <- result$regNet
top_edges <- which(abs(regNet) > 2, arr.ind = TRUE)Parameters
| Parameter | Default | Description |
|---|---|---|
gammaValue |
10 | Ratio of cells to metacells |
nPC |
25 | Number of principal components for kNN graph |
alphaValue |
0.1 | Learning rate for network updates |
hammingValue |
0.001 | Convergence threshold |
assocMethod |
“pearson” | Association method (“pearson”, “spearman”, “pcNet”) |
Performance
SCORPION v1.2.2 includes significant performance optimizations through RcppArmadillo and BLAS acceleration.
Benchmark Results
| Metric | Before Optimization | After Optimization | Speedup |
|---|---|---|---|
| Test data (230 genes × 80 cells) | ~10 sec | ~4 sec | 2.5x |
| 1000×1000 matrix multiplication | ~230 ms | 11 ms | 21x |
| Tanimoto similarity computation | ~230 ms | 19 ms | 12x |
Optimization Features
| Feature | Description |
|---|---|
| RcppArmadillo | Core Tanimoto algorithm implemented in C++ |
| BLAS Acceleration | Leverages system-optimized BLAS (OpenBLAS, Apple Accelerate, MKL) |
| Vectorization | All R operations use vectorized implementations |
| Sparse Matrix | Native support for sparse matrices via Matrix package |
Citation
If you use SCORPION in your research, please cite:
Osorio, D., Capasso, A., Eckhardt, S. G., Giri, U., Somma, A., Pitts, T. M., … & Kuijjer, M. L. (2023). Population-level comparisons of gene regulatory networks modeled on high-throughput single-cell transcriptomics data. Nature Computational Science. DOI: 10.1038/s43588-023-00558-w
Contributing
We welcome contributions! Please submit issues and pull requests on GitHub.
License
GPL-3 © Zaoqu Liu