SpaGER: Quick Start Guide
Zaoqu Liu
2026-01-25
Source:vignettes/SpaGER_tutorial.Rmd
SpaGER_tutorial.RmdIntroduction
SpaGER (Spatial Gene Expression in R) is a high-performance R implementation of the SpaGE algorithm for predicting genome-wide expression profiles in spatial transcriptomics data through integration with scRNA-seq reference datasets.
Why SpaGER?
Spatial transcriptomics technologies provide invaluable spatial context but often measure only a limited panel of genes. SpaGER addresses this limitation by:
- Leveraging scRNA-seq data to impute unmeasured genes in spatial data
- Using domain adaptation via Principal Vectors (PVs)
- Providing C++ acceleration for high performance
- Supporting seamless Seurat integration
Installation
# From R-Universe (recommended)
install.packages("SpaGER", repos = "https://zaoqu-liu.r-universe.dev")
# From GitHub
remotes::install_github("Zaoqu-Liu/SpaGER")Basic Usage
Generate Simulated Data
For demonstration, we create simulated spatial and scRNA-seq datasets:
set.seed(42)
# Simulate scRNA-seq reference data
n_rna_cells <- 500
n_spatial_cells <- 200
n_shared_genes <- 100
n_rna_only_genes <- 50
# scRNA-seq data: cells x genes
rna_data <- matrix(
abs(rnorm(n_rna_cells * (n_shared_genes + n_rna_only_genes), mean = 5, sd = 2)),
nrow = n_rna_cells
)
colnames(rna_data) <- c(
paste0("SharedGene", 1:n_shared_genes),
paste0("RNAOnlyGene", 1:n_rna_only_genes)
)
rownames(rna_data) <- paste0("RNACell", 1:n_rna_cells)
# Spatial data: only shared genes
spatial_data <- matrix(
abs(rnorm(n_spatial_cells * n_shared_genes, mean = 5, sd = 2)),
nrow = n_spatial_cells
)
colnames(spatial_data) <- paste0("SharedGene", 1:n_shared_genes)
rownames(spatial_data) <- paste0("SpatialSpot", 1:n_spatial_cells)
cat("scRNA-seq data:", nrow(rna_data), "cells x", ncol(rna_data), "genes\n")
#> scRNA-seq data: 500 cells x 150 genes
cat("Spatial data:", nrow(spatial_data), "cells x", ncol(spatial_data), "genes\n")
#> Spatial data: 200 cells x 100 genesRun SpaGE Prediction
# Predict unmeasured genes
predicted <- SpaGE(
spatial_data = as.data.frame(spatial_data),
rna_data = as.data.frame(rna_data),
n_pv = 30, # Number of principal vectors
n_neighbors = 50, # k for KNN imputation
verbose = TRUE
)
# Check results
cat("\nPredicted:", ncol(predicted), "genes for", nrow(predicted), "spatial spots\n")
#>
#> Predicted: 50 genes for 200 spatial spots
head(predicted[, 1:5])
#> RNAOnlyGene1 RNAOnlyGene2 RNAOnlyGene3 RNAOnlyGene4 RNAOnlyGene5
#> SpatialSpot1 5.052071 4.473041 5.137224 4.748652 4.786710
#> SpatialSpot2 5.204982 5.343771 5.186167 4.870106 5.362815
#> SpatialSpot3 5.500350 5.128955 4.858680 4.870086 4.983224
#> SpatialSpot4 4.914745 5.253821 4.816689 5.028372 4.672101
#> SpatialSpot5 5.061131 5.269184 4.849490 5.045723 4.868479
#> SpatialSpot6 4.912111 5.250018 5.033773 5.171062 5.024322Predict Specific Genes
# Predict only specific genes of interest
genes_of_interest <- c("RNAOnlyGene1", "RNAOnlyGene10", "RNAOnlyGene25")
predicted_specific <- SpaGE(
spatial_data = as.data.frame(spatial_data),
rna_data = as.data.frame(rna_data),
n_pv = 30,
genes_to_predict = genes_of_interest,
verbose = FALSE
)
cat("Predicted genes:", colnames(predicted_specific), "\n")
#> Predicted genes: RNAOnlyGene1 RNAOnlyGene10 RNAOnlyGene25Cross-Validation
Evaluate prediction accuracy using leave-one-gene-out cross-validation:
# Run CV on a subset of shared genes
cv_genes <- paste0("SharedGene", 1:10)
cv_results <- SpaGE_cv(
spatial_data = as.data.frame(spatial_data),
rna_data = as.data.frame(rna_data[, c(paste0("SharedGene", 1:n_shared_genes))]),
n_pv = 20,
genes = cv_genes,
verbose = FALSE
)
# Summary
cat("Cross-validation Results:\n")
#> Cross-validation Results:
cat("Mean Spearman correlation:", round(mean(cv_results$correlation), 3), "\n")
#> Mean Spearman correlation: 0.007
cat("Median Spearman correlation:", round(median(cv_results$correlation), 3), "\n")
#> Median Spearman correlation: 0.022Visualize CV Results
# Plot correlation distribution
hist(cv_results$correlation,
breaks = 20,
main = "Leave-One-Out Cross-Validation",
xlab = "Spearman Correlation",
col = "#3498db",
border = "white")
abline(v = mean(cv_results$correlation), col = "red", lwd = 2, lty = 2)
legend("topright", legend = paste("Mean =", round(mean(cv_results$correlation), 3)),
col = "red", lty = 2, lwd = 2)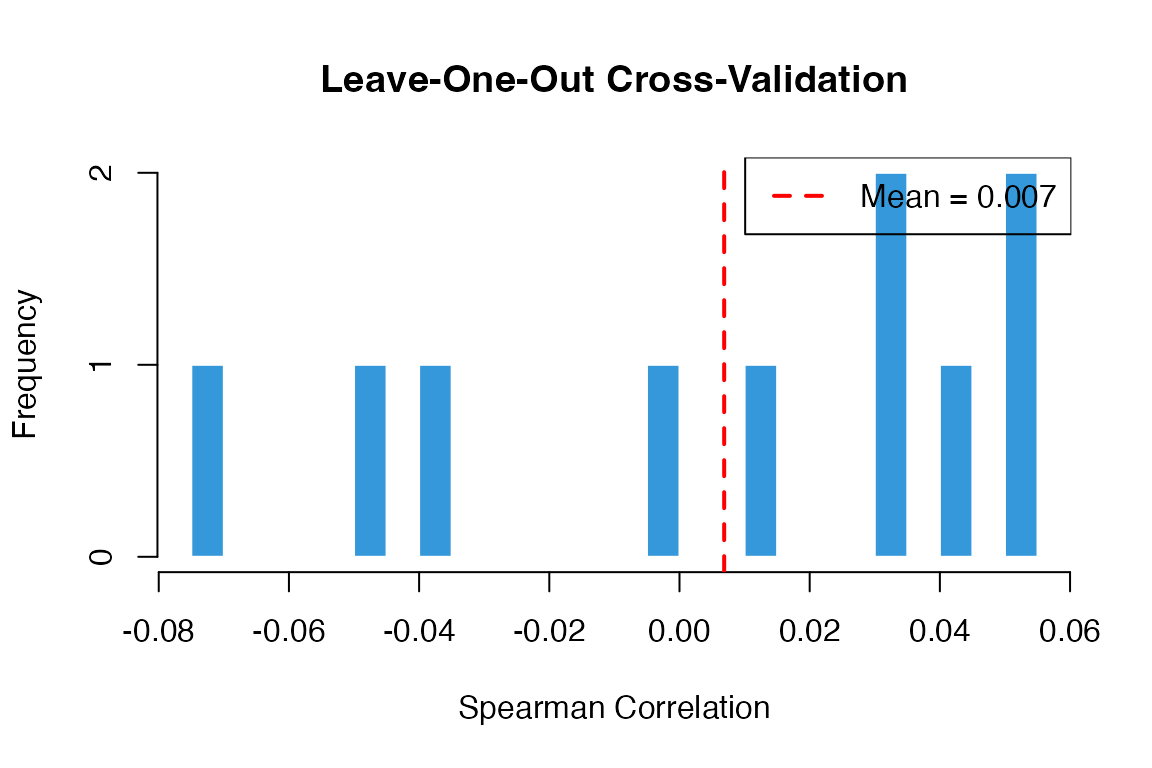
Accessing Metadata
SpaGE returns additional metadata as attributes:
# Access metadata from prediction result
cat("Number of PVs requested:", attr(predicted, "n_pv"), "\n")
#> Number of PVs requested: 30
cat("Number of PVs used:", attr(predicted, "n_pv_used"), "\n")
#> Number of PVs used: 21
cat("Number of shared genes:", attr(predicted, "n_shared_genes"), "\n")
#> Number of shared genes: 100
cat("Top PV similarities:", round(head(attr(predicted, "similarities"), 5), 3), "\n")
#> Top PV similarities: 0.893 0.86 0.838 0.823 0.798Session Information
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] SpaGER_1.0.0
#>
#> loaded via a namespace (and not attached):
#> [1] cli_3.6.5 knitr_1.51 rlang_1.1.7
#> [4] xfun_0.56 otel_0.2.0 textshaping_1.0.4
#> [7] jsonlite_2.0.0 future.apply_1.20.1 listenv_0.10.0
#> [10] htmltools_0.5.9 ragg_1.5.0 sass_0.4.10
#> [13] rmarkdown_2.30 grid_4.4.0 evaluate_1.0.5
#> [16] jquerylib_0.1.4 fastmap_1.2.0 yaml_2.3.12
#> [19] lifecycle_1.0.5 FNN_1.1.4.1 compiler_4.4.0
#> [22] codetools_0.2-20 irlba_2.3.5.1 fs_1.6.6
#> [25] Rcpp_1.1.1 htmlwidgets_1.6.4 future_1.69.0
#> [28] lattice_0.22-7 systemfonts_1.3.1 digest_0.6.39
#> [31] R6_2.6.1 parallelly_1.46.1 parallel_4.4.0
#> [34] bslib_0.9.0 Matrix_1.7-4 tools_4.4.0
#> [37] globals_0.18.0 pkgdown_2.1.3 cachem_1.1.0
#> [40] desc_1.4.3