SpaGER: Algorithm and Mathematical Foundation
Zaoqu Liu
2026-01-25
Source:vignettes/algorithm.Rmd
algorithm.RmdOverview
SpaGER implements the SpaGE (Spatial Gene Enhancement) algorithm, which uses domain adaptation to predict unmeasured gene expression in spatial transcriptomics data by leveraging scRNA-seq reference data.
The Challenge
Spatial transcriptomics technologies face a fundamental trade-off:
| Technology | Genes Measured | Spatial Resolution |
|---|---|---|
| MERFISH | ~100-1000 | Subcellular |
| seqFISH | ~100-10000 | Subcellular |
| Visium | ~20000 | ~55μm spots |
| Slide-seq | ~20000 | ~10μm beads |
High-resolution methods (MERFISH, seqFISH) measure fewer genes, while whole-transcriptome methods (Visium) have lower spatial resolution. SpaGER bridges this gap by using scRNA-seq to predict unmeasured genes.
Algorithm Steps
Step 1: Data Standardization
Both datasets are z-score normalized using population standard deviation:
where: - is the expression of gene in cell - is the mean expression - is the population standard deviation
library(SpaGER)
# Demonstrate z-score standardization
set.seed(42)
X <- matrix(c(1, 2, 3, 4, 5, 6, 7, 8, 9), nrow = 3)
X_std <- zscore(X)
cat("Original data:\n")
#> Original data:
print(X)
#> [,1] [,2] [,3]
#> [1,] 1 4 7
#> [2,] 2 5 8
#> [3,] 3 6 9
cat("\nStandardized data:\n")
#>
#> Standardized data:
print(round(X_std, 4))
#> [,1] [,2] [,3]
#> [1,] -1.2247 -1.2247 -1.2247
#> [2,] 0.0000 0.0000 0.0000
#> [3,] 1.2247 1.2247 1.2247
cat("\nColumn means (should be ~0):", round(colMeans(X_std), 10), "\n")
#>
#> Column means (should be ~0): 0 0 0Step 2: Dimensionality Reduction
Both datasets are independently reduced to a lower-dimensional space using PCA (or other methods):
where contains the principal component loadings.
set.seed(42)
# Create sample data with structure
n <- 100
X_source <- cbind(
rnorm(n, 0, 3), # High variance
rnorm(n, 0, 1), # Medium variance
rnorm(n, 0, 0.3) # Low variance
)
# Perform PCA
reducer <- process_dim_reduction("pca", n_dim = 2)
reducer <- reducer$fit(X_source)
cat("PCA loadings (components):\n")
#> PCA loadings (components):
print(round(reducer$components_, 4))
#> [,1] [,2] [,3]
#> PC1 -0.9999 -0.0098 0.0142
#> PC2 -0.0094 0.9995 0.0289
cat("\nFirst PC captures high-variance direction\n")
#>
#> First PC captures high-variance directionStep 3: Matrix Orthogonalization
The factor matrices are orthonormalized using SVD:
This ensures the basis vectors are orthogonal and unit-length.
Step 4: Principal Vectors Computation
The key innovation of SpaGE is finding Principal Vectors (PVs) that represent shared variation between domains.
Given orthonormalized factor matrices (source/scRNA-seq) and (target/spatial), we compute:
The singular values represent the cosine similarity between the -th pair of principal vectors. Higher values indicate better alignment.
set.seed(42)
# Create two datasets with shared structure
n_samples <- 100
n_features <- 50
# Shared signal
shared <- matrix(rnorm(n_samples * 10), nrow = n_samples)
# Source and target with shared structure
source_data <- cbind(
shared %*% matrix(rnorm(10 * n_features), nrow = 10),
matrix(rnorm(n_samples * n_features, sd = 0.5), nrow = n_samples)
)[, 1:n_features]
target_data <- cbind(
shared %*% matrix(rnorm(10 * n_features), nrow = 10) + matrix(rnorm(n_samples * n_features, sd = 0.3), nrow = n_samples),
matrix(rnorm(n_samples * n_features, sd = 0.5), nrow = n_samples)
)[, 1:n_features]
# Compute principal vectors
pv <- PVComputation(n_factors = 20, n_pv = 20, dim_reduction = "pca")
pv <- pv$fit(source_data, target_data)
# Plot cosine similarities
similarities <- diag(pv$cosine_similarity_matrix_)
barplot(similarities,
names.arg = 1:length(similarities),
main = "Principal Vector Cosine Similarities",
xlab = "Principal Vector Index",
ylab = "Cosine Similarity",
col = ifelse(similarities > 0.3, "#2ecc71", "#e74c3c"),
border = "white")
abline(h = 0.3, lty = 2, col = "blue", lwd = 2)
legend("topright",
legend = c("Selected (>0.3)", "Rejected (≤0.3)", "Threshold"),
fill = c("#2ecc71", "#e74c3c", NA),
border = c("white", "white", NA),
lty = c(NA, NA, 2),
col = c(NA, NA, "blue"))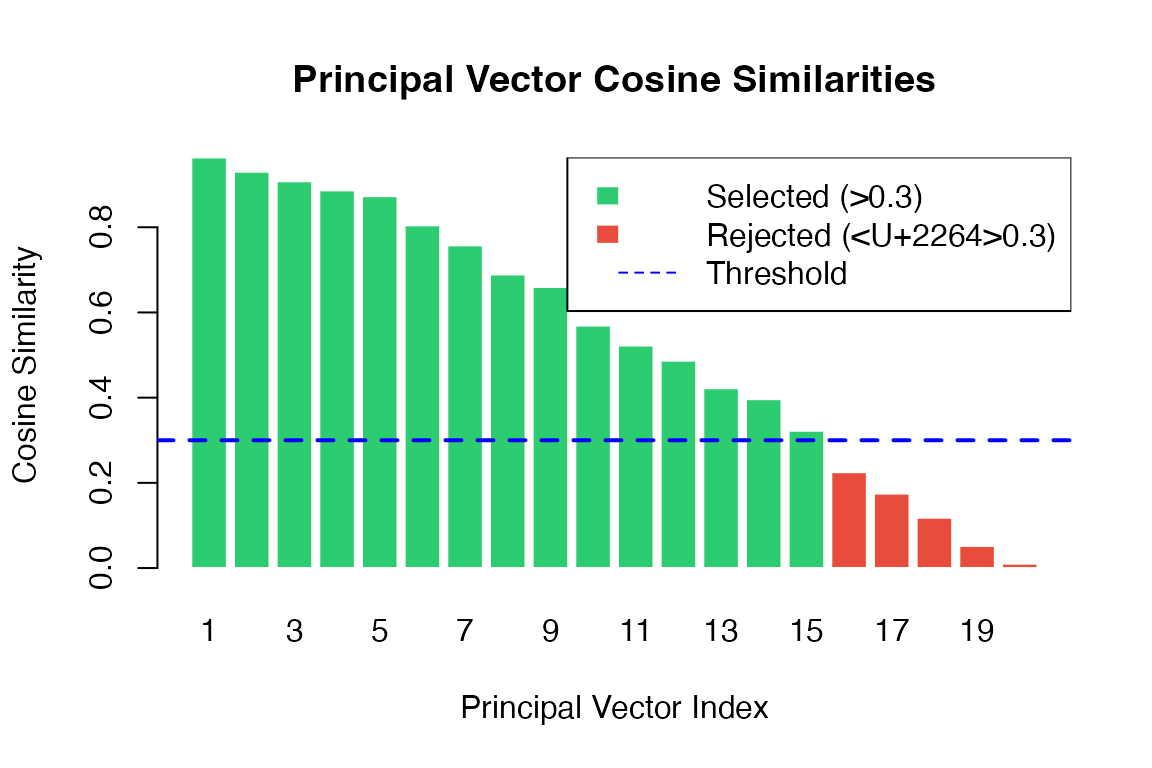
Step 5: Projection
Both datasets are projected onto the aligned subspace defined by the selected principal vectors:
where contains the source principal vector loadings.
Step 6: Weighted k-NN Imputation
For each spatial cell , find nearest scRNA-seq cells using cosine distance:
Expression is predicted as a weighted average:
where weights are computed as:
set.seed(42)
# Demonstrate KNN with cosine distance
query <- matrix(c(1, 0), nrow = 1)
ref <- matrix(c(
0.9, 0.1, # Close to query
0.7, 0.3, # Medium distance
0.5, 0.5, # Farther
0.1, 0.9 # Far
), nrow = 4, byrow = TRUE)
# Compute KNN
knn_result <- cosine_knn(query, ref, k = 3)
# Visualize
plot(ref[,1], ref[,2], pch = 19, cex = 2,
xlim = c(-0.2, 1.2), ylim = c(-0.2, 1.2),
xlab = "Dimension 1", ylab = "Dimension 2",
main = "Cosine KNN Illustration",
col = c("#2ecc71", "#f39c12", "#e74c3c", "#95a5a6"))
points(query[1,1], query[1,2], pch = 18, cex = 3, col = "#3498db")
# Draw connections to neighbors
for (i in 1:3) {
idx <- knn_result$indices[1, i]
lines(c(query[1,1], ref[idx,1]), c(query[1,2], ref[idx,2]),
lty = 2, col = "#3498db", lwd = 1.5)
}
legend("topright",
legend = c("Query", "NN 1", "NN 2", "NN 3", "Not selected"),
pch = c(18, 19, 19, 19, 19),
col = c("#3498db", "#2ecc71", "#f39c12", "#e74c3c", "#95a5a6"),
pt.cex = c(2, 1.5, 1.5, 1.5, 1.5))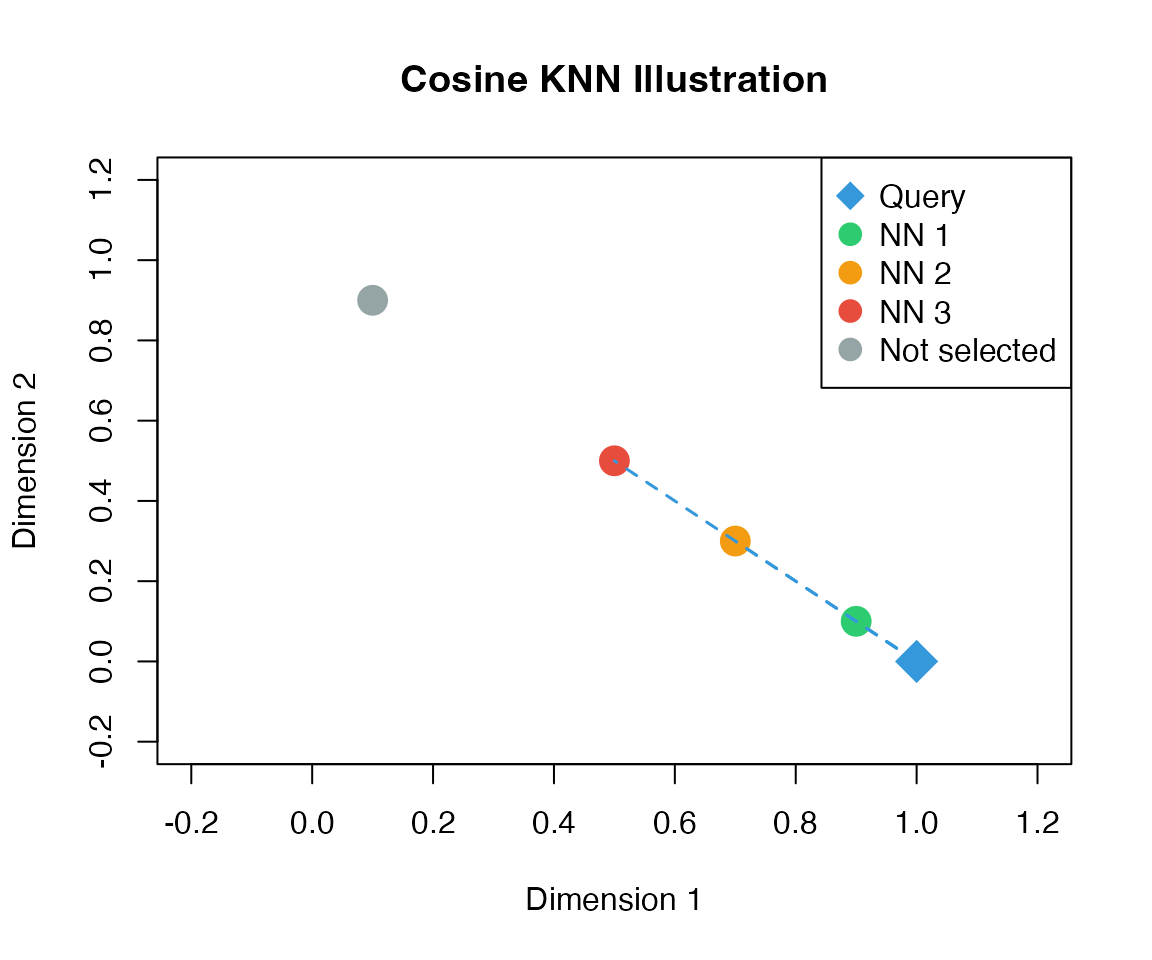
Comparison with Python Implementation
SpaGER produces numerically equivalent results to the original Python SpaGE:
set.seed(42)
# Z-score comparison
X <- matrix(rnorm(30), nrow = 10, ncol = 3)
X_r <- zscore(X)
# Manual calculation (matching scipy.stats.zscore)
X_manual <- scale(X, center = TRUE, scale = TRUE) * sqrt(nrow(X) / (nrow(X) - 1))
cat("Maximum difference from Python-equivalent calculation:",
max(abs(X_r - X_manual)), "\n")
#> Maximum difference from Python-equivalent calculation: 4.440892e-16Summary
The SpaGE algorithm workflow:
┌─────────────────┐ ┌─────────────────┐
│ Spatial Data │ │ scRNA-seq Data │
│ (limited genes)│ │ (full genome) │
└────────┬────────┘ └────────┬────────┘
│ │
▼ ▼
┌────────────────────────────────┐
│ Z-score Standardization │
└────────────────────────────────┘
│ │
▼ ▼
┌──────────┐ ┌──────────┐
│ PCA │ │ PCA │
└────┬─────┘ └────┬─────┘
│ │
└───────────┬───────────┘
▼
┌────────────────────────────────┐
│ Principal Vectors (SVD) │
│ Select PVs with sim > 0.3 │
└────────────────────────────────┘
│
▼
┌────────────────────────────────┐
│ Project onto aligned space │
└────────────────────────────────┘
│
▼
┌────────────────────────────────┐
│ Weighted k-NN Imputation │
│ (cosine distance) │
└────────────────────────────────┘
│
▼
┌────────────────────────────────┐
│ Predicted Gene Expression │
└────────────────────────────────┘References
Abdelaal T, et al. (2020). SpaGE: Spatial Gene Enhancement using scRNA-seq. Nucleic Acids Research, 48(18):e107.
Mourragui S, et al. (2019). PRECISE: A domain adaptation approach to transfer predictors of drug response from pre-clinical models to tumors. Bioinformatics, 35(14):i510-i519.
Session Information
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] SpaGER_1.0.0
#>
#> loaded via a namespace (and not attached):
#> [1] cli_3.6.5 knitr_1.51 rlang_1.1.7
#> [4] xfun_0.56 otel_0.2.0 textshaping_1.0.4
#> [7] jsonlite_2.0.0 future.apply_1.20.1 listenv_0.10.0
#> [10] htmltools_0.5.9 ragg_1.5.0 sass_0.4.10
#> [13] rmarkdown_2.30 grid_4.4.0 evaluate_1.0.5
#> [16] jquerylib_0.1.4 fastmap_1.2.0 yaml_2.3.12
#> [19] lifecycle_1.0.5 FNN_1.1.4.1 compiler_4.4.0
#> [22] codetools_0.2-20 irlba_2.3.5.1 fs_1.6.6
#> [25] Rcpp_1.1.1 htmlwidgets_1.6.4 future_1.69.0
#> [28] lattice_0.22-7 systemfonts_1.3.1 digest_0.6.39
#> [31] R6_2.6.1 parallelly_1.46.1 parallel_4.4.0
#> [34] bslib_0.9.0 Matrix_1.7-4 tools_4.4.0
#> [37] globals_0.18.0 pkgdown_2.1.3 cachem_1.1.0
#> [40] desc_1.4.3