SpaGER: Visualization and Analysis
Zaoqu Liu
2026-01-25
Source:vignettes/visualization.Rmd
visualization.RmdIntroduction
This vignette demonstrates visualization techniques for SpaGER predictions and analyses, helping you interpret and present your results effectively.
Simulated Dataset
We create a more realistic simulated dataset with biological structure:
# Parameters
n_rna <- 500
n_spatial <- 200
n_shared <- 80
n_predict <- 40
# Create cell type structure (3 cell types)
cell_types <- rep(1:3, length.out = n_rna)
type_means <- c(3, 5, 7)
# scRNA-seq with cell type-specific expression
rna_base <- matrix(0, nrow = n_rna, ncol = n_shared + n_predict)
for (i in 1:n_rna) {
rna_base[i, ] <- rnorm(n_shared + n_predict, mean = type_means[cell_types[i]], sd = 1)
}
rna_base <- abs(rna_base)
colnames(rna_base) <- c(paste0("Shared", 1:n_shared), paste0("Novel", 1:n_predict))
# Spatial data - mixture of cell types
spatial_types <- sample(1:3, n_spatial, replace = TRUE, prob = c(0.5, 0.3, 0.2))
spatial_base <- matrix(0, nrow = n_spatial, ncol = n_shared)
for (i in 1:n_spatial) {
spatial_base[i, ] <- rnorm(n_shared, mean = type_means[spatial_types[i]], sd = 1.2)
}
spatial_base <- abs(spatial_base)
colnames(spatial_base) <- paste0("Shared", 1:n_shared)
# Add spatial coordinates
coords <- data.frame(
x = runif(n_spatial, 0, 100),
y = runif(n_spatial, 0, 100)
)
rownames(coords) <- paste0("Spot", 1:n_spatial)
rownames(spatial_base) <- rownames(coords)
cat("Data created:\n")
#> Data created:
cat(" scRNA-seq:", n_rna, "cells,", ncol(rna_base), "genes\n")
#> scRNA-seq: 500 cells, 120 genes
cat(" Spatial:", n_spatial, "spots,", ncol(spatial_base), "genes\n")
#> Spatial: 200 spots, 80 genesRun SpaGE Prediction
predicted <- SpaGE(
spatial_data = as.data.frame(spatial_base),
rna_data = as.data.frame(rna_base),
n_pv = 30,
n_neighbors = 50,
verbose = FALSE
)
cat("Predicted", ncol(predicted), "novel genes\n")
#> Predicted 40 novel genesVisualizing Principal Vector Selection
# Get PV information
similarities <- attr(predicted, "similarities")
n_pv_used <- attr(predicted, "n_pv_used")
par(mfrow = c(1, 2))
# Bar plot of similarities
cols <- ifelse(seq_along(similarities) <= n_pv_used, "#3498db", "#bdc3c7")
barplot(similarities,
names.arg = seq_along(similarities),
col = cols,
border = "white",
main = "Principal Vector Selection",
xlab = "PV Index",
ylab = "Cosine Similarity",
ylim = c(0, 1))
abline(h = 0.3, col = "#e74c3c", lty = 2, lwd = 2)
legend("topright",
legend = c("Selected", "Rejected", "Threshold (0.3)"),
fill = c("#3498db", "#bdc3c7", NA),
border = c("white", "white", NA),
lty = c(NA, NA, 2),
col = c(NA, NA, "#e74c3c"))
# Cumulative variance
cum_var <- cumsum(similarities^2) / sum(similarities^2)
plot(cum_var, type = "b", pch = 19, col = "#2ecc71",
main = "Cumulative Variance Captured",
xlab = "Number of PVs",
ylab = "Cumulative Proportion",
xlim = c(1, length(similarities)))
abline(v = n_pv_used, col = "#e74c3c", lty = 2)
abline(h = cum_var[n_pv_used], col = "#e74c3c", lty = 2)
text(n_pv_used + 1, cum_var[n_pv_used],
sprintf("%.1f%%", cum_var[n_pv_used] * 100), pos = 4)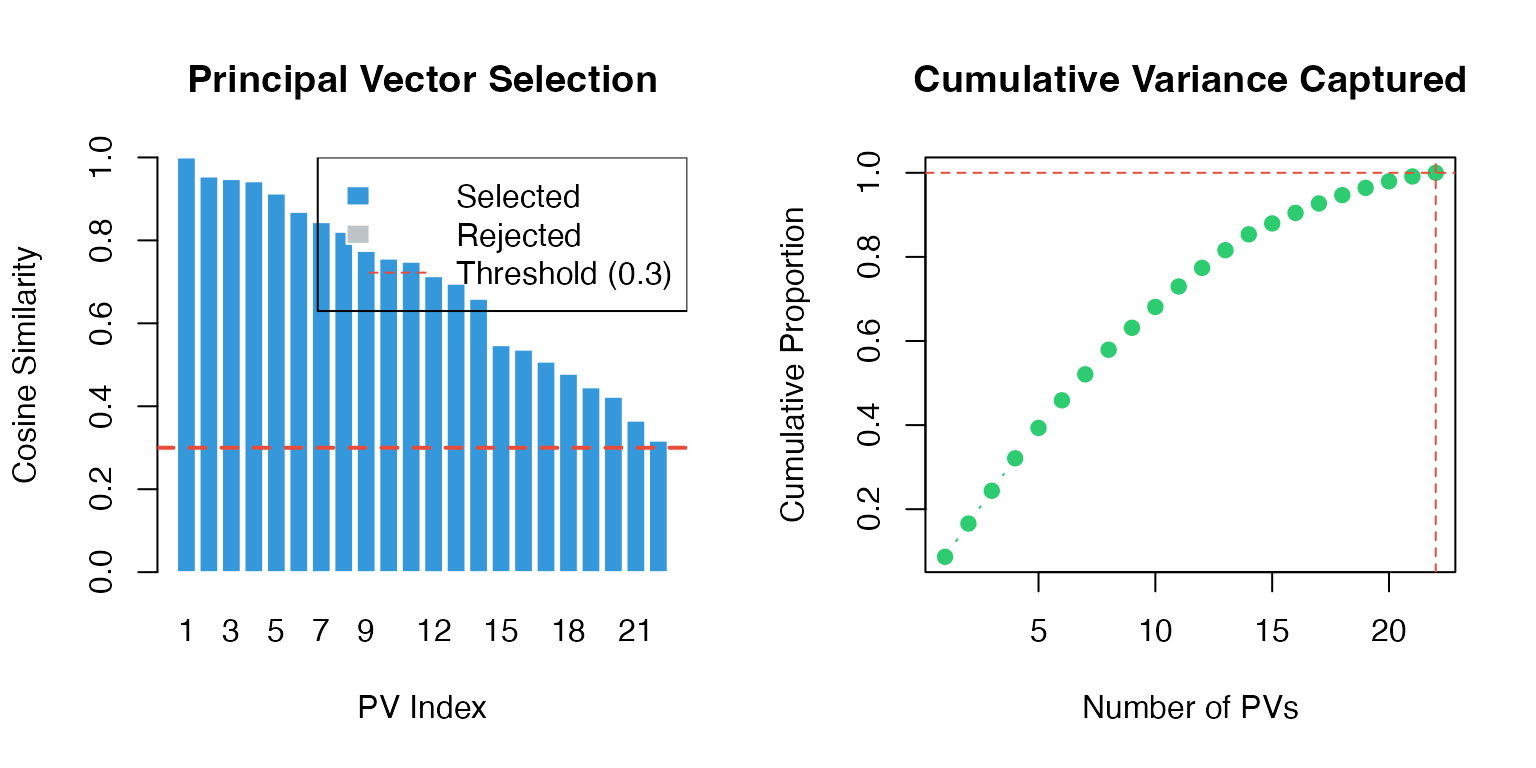
Spatial Expression Patterns
# Function to plot spatial expression
plot_spatial <- function(coords, values, title, col_palette = NULL) {
if (is.null(col_palette)) {
col_palette <- colorRampPalette(c("#313695", "#4575b4", "#74add1",
"#abd9e9", "#ffffbf", "#fee090",
"#fdae61", "#f46d43", "#d73027"))(100)
}
# Normalize values to [0, 1]
val_norm <- (values - min(values)) / (max(values) - min(values) + 1e-10)
cols <- col_palette[ceiling(val_norm * 99) + 1]
plot(coords$x, coords$y, pch = 19, cex = 1.5,
col = cols, xlab = "X", ylab = "Y", main = title,
asp = 1)
}
# Plot predicted genes
par(mfrow = c(2, 2), mar = c(4, 4, 3, 1))
for (i in 1:4) {
gene <- colnames(predicted)[i]
plot_spatial(coords, predicted[, gene], paste("Predicted:", gene))
}
Cross-Validation Results
# Run CV
cv_results <- SpaGE_cv(
spatial_data = as.data.frame(spatial_base),
rna_data = as.data.frame(rna_base[, paste0("Shared", 1:n_shared)]),
n_pv = 20,
genes = paste0("Shared", 1:20),
verbose = FALSE
)
par(mfrow = c(1, 2))
# Histogram
hist(cv_results$correlation, breaks = 15, col = "#3498db", border = "white",
main = "Cross-Validation Performance",
xlab = "Spearman Correlation",
xlim = c(-0.2, 1))
abline(v = median(cv_results$correlation), col = "#e74c3c", lwd = 2, lty = 2)
abline(v = mean(cv_results$correlation), col = "#2ecc71", lwd = 2, lty = 2)
legend("topleft",
legend = c(paste("Median:", round(median(cv_results$correlation), 3)),
paste("Mean:", round(mean(cv_results$correlation), 3))),
col = c("#e74c3c", "#2ecc71"), lty = 2, lwd = 2)
# Per-gene plot
cv_ordered <- cv_results[order(cv_results$correlation, decreasing = TRUE), ]
barplot(cv_ordered$correlation,
names.arg = substr(cv_ordered$gene, 7, 10),
las = 2,
col = ifelse(cv_ordered$correlation > 0.3, "#2ecc71", "#e74c3c"),
border = "white",
main = "Per-Gene Correlation",
ylab = "Spearman Correlation")
abline(h = 0.3, lty = 2)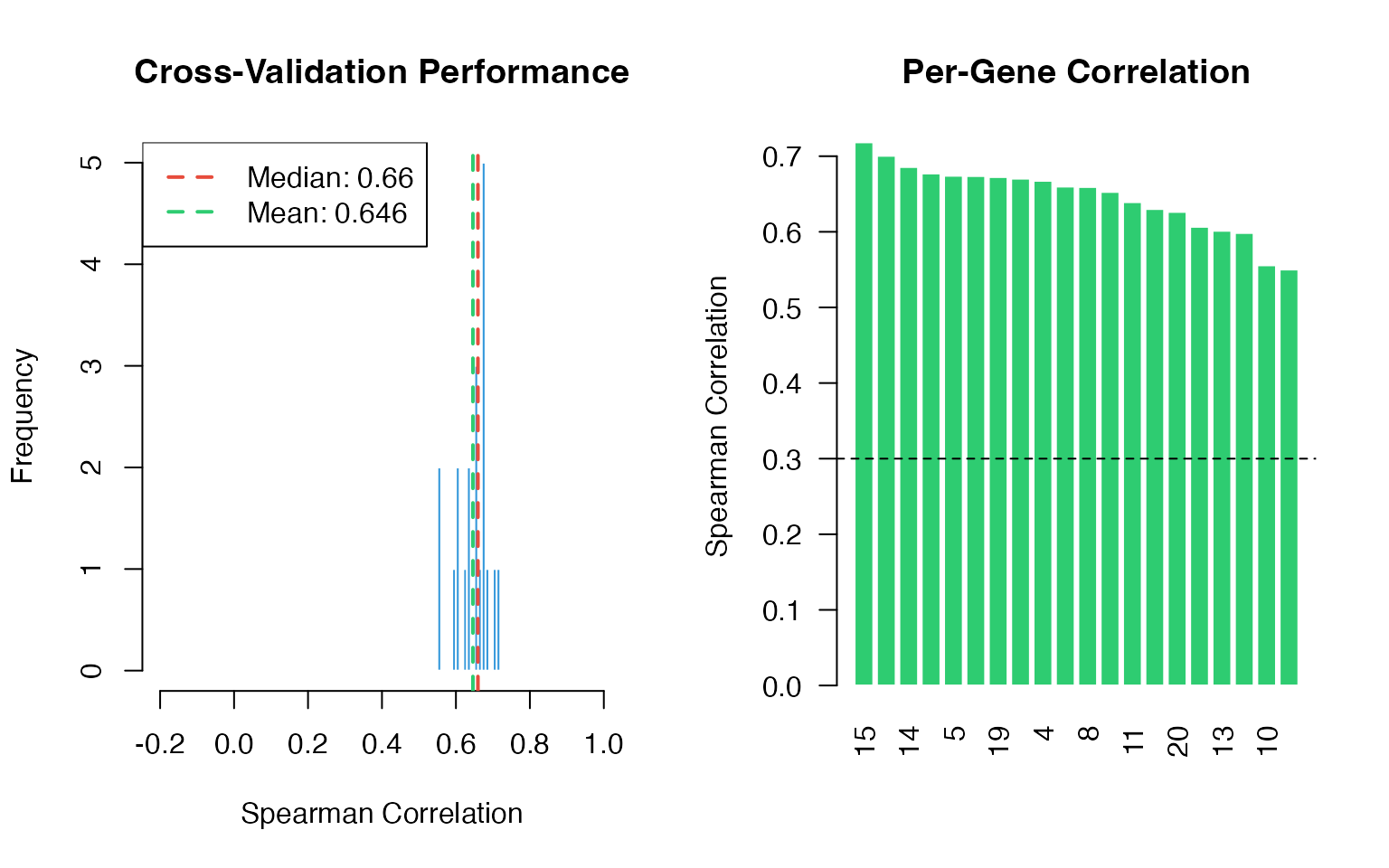
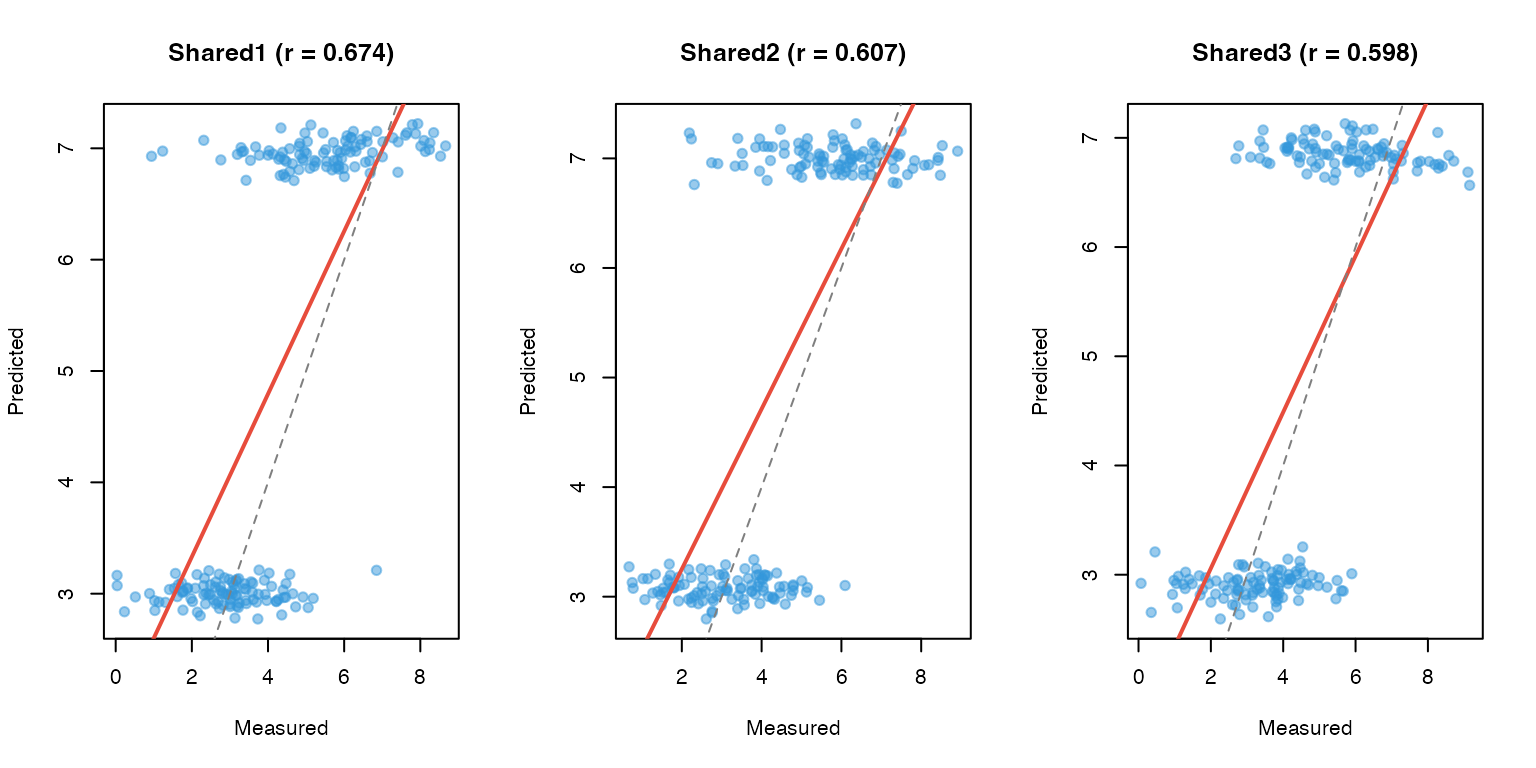
Measured vs Predicted Scatter Plots
# For CV genes, compare measured vs predicted
par(mfrow = c(1, 3))
for (i in 1:3) {
gene <- cv_results$gene[i]
# Get measured values
measured <- spatial_base[, gene]
# Run prediction for this gene
spatial_minus <- spatial_base[, setdiff(colnames(spatial_base), gene)]
pred_single <- SpaGE(
spatial_data = as.data.frame(spatial_minus),
rna_data = as.data.frame(rna_base[, paste0("Shared", 1:n_shared)]),
n_pv = 20,
genes_to_predict = gene,
verbose = FALSE
)
predicted_vals <- pred_single[, gene]
cor_val <- cor(measured, predicted_vals, method = "spearman")
plot(measured, predicted_vals, pch = 19, col = adjustcolor("#3498db", 0.5),
xlab = "Measured", ylab = "Predicted",
main = sprintf("%s (r = %.3f)", gene, cor_val))
abline(lm(predicted_vals ~ measured), col = "#e74c3c", lwd = 2)
# Add identity line
abline(0, 1, lty = 2, col = "gray50")
}
Expression Distribution Comparison
par(mfrow = c(1, 2))
# Density plot for predicted genes
gene1 <- colnames(predicted)[1]
gene2 <- colnames(predicted)[2]
# Use density estimation
d1 <- density(predicted[, gene1])
d2 <- density(predicted[, gene2])
plot(d1, col = "#3498db", lwd = 2,
main = "Predicted Expression Distribution",
xlab = "Expression Level",
xlim = range(c(d1$x, d2$x)),
ylim = range(c(d1$y, d2$y)))
lines(d2, col = "#e74c3c", lwd = 2)
legend("topright", legend = c(gene1, gene2),
col = c("#3498db", "#e74c3c"), lwd = 2)
# Box plot
boxplot(predicted[, 1:10],
col = "#3498db",
border = "#2c3e50",
main = "Predicted Expression (First 10 Genes)",
xlab = "Gene",
ylab = "Expression",
las = 2,
names = substr(colnames(predicted)[1:10], 1, 8))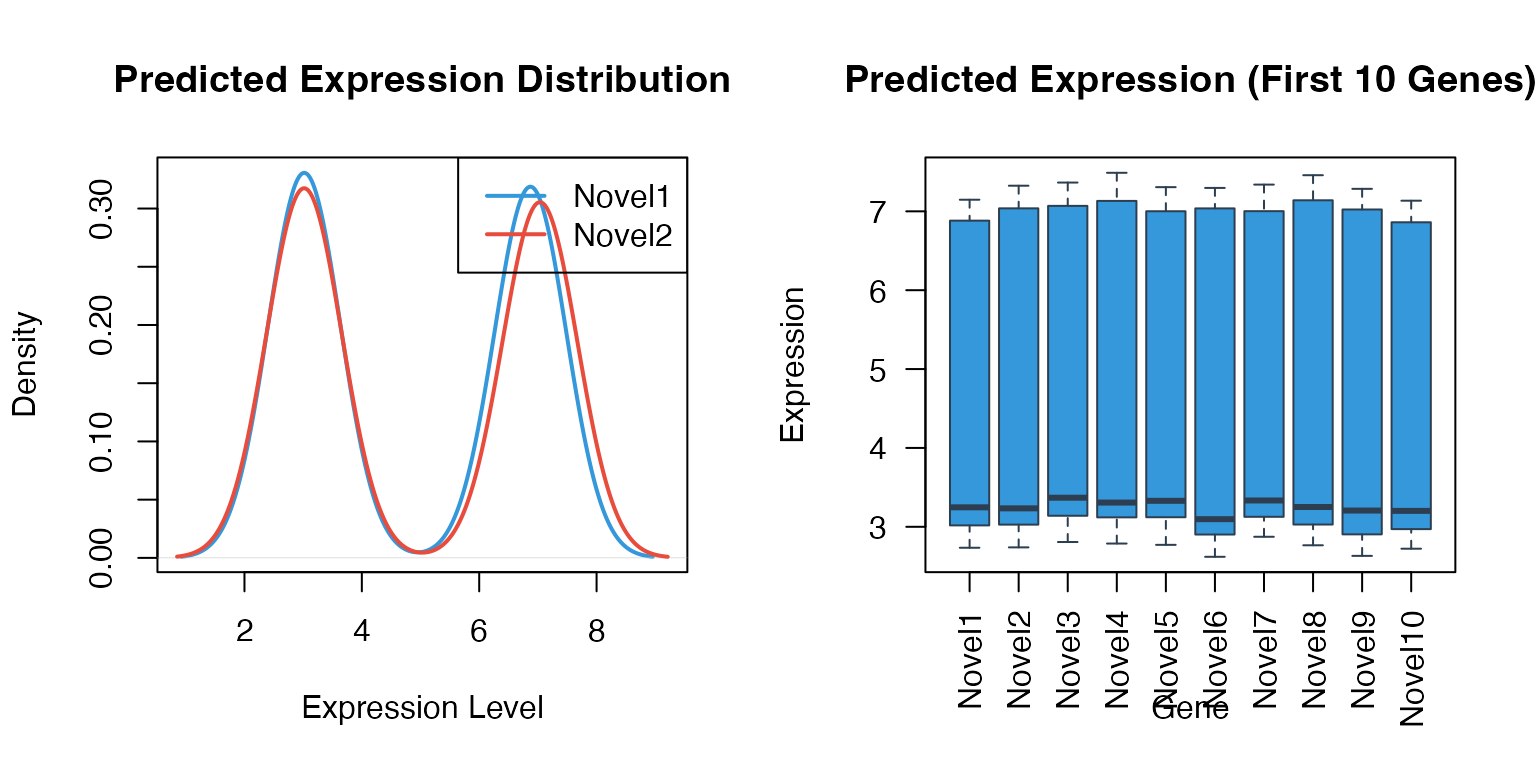
Correlation Heatmap
# Compute correlation matrix for predicted genes
pred_subset <- predicted[, 1:15]
cor_mat <- cor(pred_subset, method = "spearman")
# Simple heatmap
heatmap(cor_mat,
col = colorRampPalette(c("#3498db", "white", "#e74c3c"))(50),
scale = "none",
main = "Gene-Gene Correlation (Predicted)",
margins = c(8, 8))
Summary Statistics
cat("=== SpaGE Prediction Summary ===\n\n")
#> === SpaGE Prediction Summary ===
cat("Input Data:\n")
#> Input Data:
cat(sprintf(" Spatial spots: %d\n", nrow(spatial_base)))
#> Spatial spots: 200
cat(sprintf(" scRNA-seq cells: %d\n", nrow(rna_base)))
#> scRNA-seq cells: 500
cat(sprintf(" Shared genes: %d\n", n_shared))
#> Shared genes: 80
cat(sprintf(" Novel genes predicted: %d\n", ncol(predicted)))
#> Novel genes predicted: 40
cat("\nPrincipal Vectors:\n")
#>
#> Principal Vectors:
cat(sprintf(" Requested: %d\n", attr(predicted, "n_pv")))
#> Requested: 30
cat(sprintf(" Used (sim > 0.3): %d\n", attr(predicted, "n_pv_used")))
#> Used (sim > 0.3): 22
cat(sprintf(" Mean similarity: %.3f\n", mean(attr(predicted, "similarities"))))
#> Mean similarity: 0.694
cat("\nCross-Validation:\n")
#>
#> Cross-Validation:
cat(sprintf(" Genes tested: %d\n", nrow(cv_results)))
#> Genes tested: 20
cat(sprintf(" Mean correlation: %.3f\n", mean(cv_results$correlation)))
#> Mean correlation: 0.646
cat(sprintf(" Median correlation: %.3f\n", median(cv_results$correlation)))
#> Median correlation: 0.660
cat(sprintf(" Genes with r > 0.3: %d (%.1f%%)\n",
sum(cv_results$correlation > 0.3),
100 * mean(cv_results$correlation > 0.3)))
#> Genes with r > 0.3: 20 (100.0%)
cat("\nPredicted Expression:\n")
#>
#> Predicted Expression:
cat(sprintf(" Mean: %.3f\n", mean(as.matrix(predicted))))
#> Mean: 4.962
cat(sprintf(" SD: %.3f\n", sd(as.matrix(predicted))))
#> SD: 1.997
cat(sprintf(" Range: [%.3f, %.3f]\n", min(predicted), max(predicted)))
#> Range: [2.564, 7.563]Exporting Results
# Export predicted expression
write.csv(predicted, "spage_predictions.csv")
# Export CV results
write.csv(cv_results, "spage_cv_results.csv")
# Export for visualization in other tools
saveRDS(list(
predictions = predicted,
coordinates = coords,
cv_results = cv_results,
metadata = list(
n_pv = attr(predicted, "n_pv"),
n_pv_used = attr(predicted, "n_pv_used"),
similarities = attr(predicted, "similarities")
)
), "spage_results.rds")Session Information
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] SpaGER_1.0.0
#>
#> loaded via a namespace (and not attached):
#> [1] cli_3.6.5 knitr_1.51 rlang_1.1.7
#> [4] xfun_0.56 otel_0.2.0 textshaping_1.0.4
#> [7] jsonlite_2.0.0 future.apply_1.20.1 listenv_0.10.0
#> [10] htmltools_0.5.9 ragg_1.5.0 sass_0.4.10
#> [13] rmarkdown_2.30 grid_4.4.0 evaluate_1.0.5
#> [16] jquerylib_0.1.4 fastmap_1.2.0 yaml_2.3.12
#> [19] lifecycle_1.0.5 FNN_1.1.4.1 compiler_4.4.0
#> [22] codetools_0.2-20 irlba_2.3.5.1 fs_1.6.6
#> [25] Rcpp_1.1.1 htmlwidgets_1.6.4 future_1.69.0
#> [28] lattice_0.22-7 systemfonts_1.3.1 digest_0.6.39
#> [31] R6_2.6.1 parallelly_1.46.1 parallel_4.4.0
#> [34] bslib_0.9.0 Matrix_1.7-4 tools_4.4.0
#> [37] globals_0.18.0 pkgdown_2.1.3 cachem_1.1.0
#> [40] desc_1.4.3