Overview
fastCNV is an R package developed for efficient and accurate inference of Copy Number Variations (CNVs) from single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics data. It provides a comprehensive framework for:
- Detecting chromosomal amplifications and deletions
- Identifying tumor subclones based on CNV profiles
- Reconstructing clonal phylogenetic trees
- Generating publication-ready visualizations
Installation
From R-universe (Recommended)
install.packages("fastCNV", repos = "https://zaoqu-liu.r-universe.dev")From GitHub
if (!requireNamespace("remotes", quietly = TRUE)) {
install.packages("remotes")
}
remotes::install_github("Zaoqu-Liu/fastCNV")Quick Start
Basic Workflow
The main function fastCNV() provides an all-in-one
workflow:
library(fastCNV)
# Load your Seurat object
# seurat_obj <- readRDS("your_seurat_object.rds")
# Run CNV analysis
result <- fastCNV(
seuratObj = seurat_obj,
sampleName = "Sample1",
referenceVar = "cell_type",
referenceLabel = c("Normal_epithelial", "Fibroblast"),
prepareCounts = TRUE,
getCNVPerChromosomeArm = TRUE,
getCNVClusters = TRUE,
doPlot = TRUE
)Understanding the Parameters
| Parameter | Description | Default |
|---|---|---|
seuratObj |
Seurat object or list of Seurat objects | Required |
sampleName |
Sample identifier(s) | Required |
referenceVar |
Metadata column for cell type annotation | Required |
referenceLabel |
Cell types to use as reference | Required |
windowSize |
Number of genes per sliding window | 150 |
topNGenes |
Top expressed genes to consider | 7000 |
prepareCounts |
Aggregate counts (for spatial data) | TRUE |
aggregFactor |
Aggregation factor for spatial data | 10 |
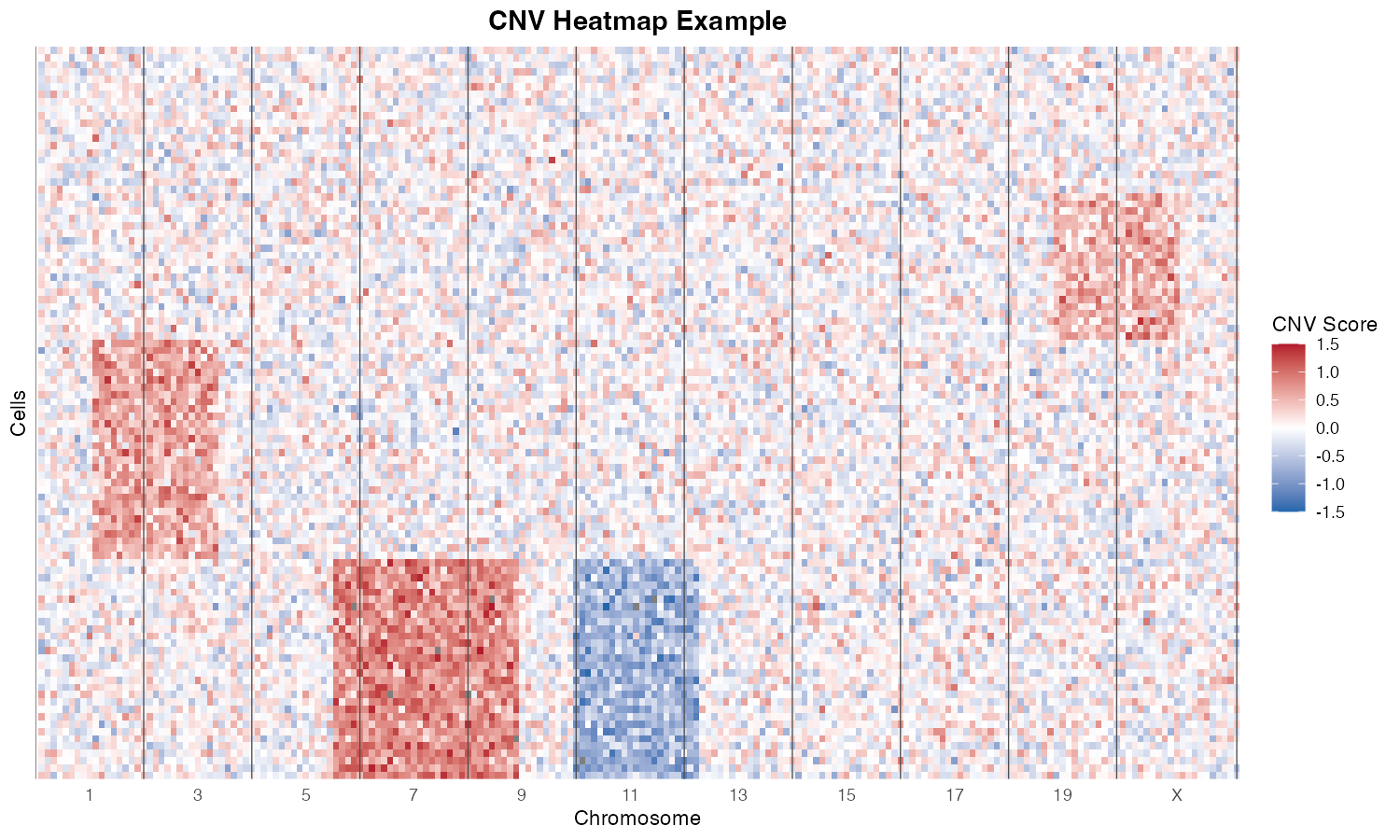
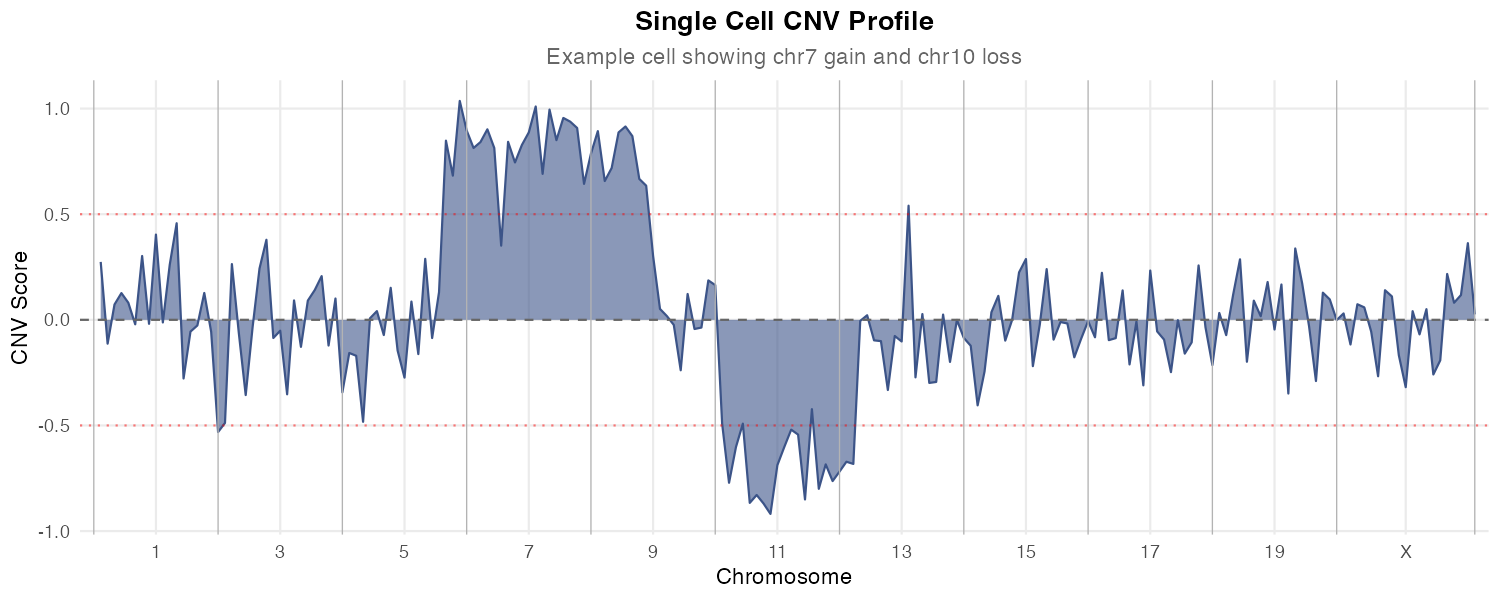
Examining Results
After running fastCNV(), the results are stored in the
Seurat object’s metadata:
Supported Data Types
10X Visium HD
result <- fastCNV_10XHD(
seuratObjHD = visium_hd_seurat,
sampleName = "VisiumHD_sample",
referenceVar = "region",
referenceLabel = c("Normal_tissue"),
doPlot = TRUE
)Next Steps
- See the Algorithm Methodology vignette for detailed explanations
- Explore Advanced Visualization options
- Learn about Multi-Sample Analysis workflows
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> loaded via a namespace (and not attached):
#> [1] digest_0.6.39 desc_1.4.3 R6_2.6.1 fastmap_1.2.0
#> [5] xfun_0.56 cachem_1.1.0 knitr_1.51 htmltools_0.5.9
#> [9] rmarkdown_2.30 lifecycle_1.0.5 cli_3.6.5 sass_0.4.10
#> [13] pkgdown_2.1.3 textshaping_1.0.4 jquerylib_0.1.4 systemfonts_1.3.1
#> [17] compiler_4.4.0 tools_4.4.0 ragg_1.5.0 bslib_0.9.0
#> [21] evaluate_1.0.5 yaml_2.3.12 otel_0.2.0 jsonlite_2.0.0
#> [25] rlang_1.1.7 fs_1.6.6 htmlwidgets_1.6.4