Overview
fastCNV provides comprehensive visualization tools for CNV analysis results. This vignette covers:
- CNV Heatmaps - Genome-wide CNV profiles
- Phylogenetic Trees - Clonal evolution visualization
- Chromosome Arm Plots - Arm-level CNV summary
- Spatial CNV Maps - CNV patterns in tissue context
CNV Heatmaps
Example CNV Heatmap
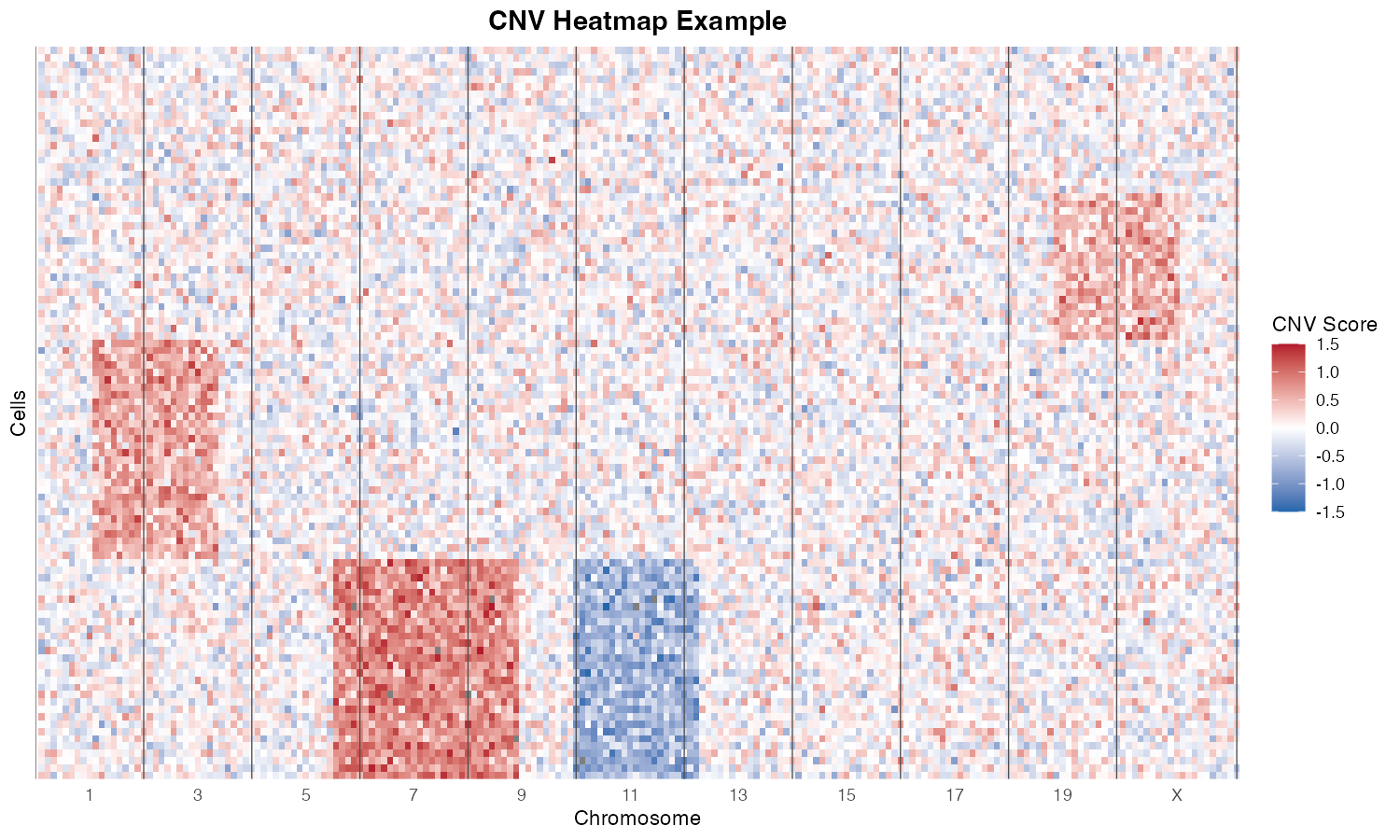
CNV heatmap showing chromosomal alterations. Rows represent cells, columns represent genomic windows. Blue = deletion, Red = amplification.
Basic Heatmap
The plotCNVResults() function generates
publication-ready heatmaps:
library(fastCNV)
# Generate CNV heatmap
plotCNVResults(
seuratObj = result,
referenceVar = "cell_type",
tumorLabel = "Tumor"
)Customizing Heatmaps
Split by Cell Type
plotCNVResults(
seuratObj = result,
referenceVar = "cell_type",
tumorLabel = "Tumor",
splitPlotOnVar = "cell_subtype" # Split rows by subtype
)Split by CNV Clusters
plotCNVResults(
seuratObj = result,
referenceVar = "cell_type",
tumorLabel = "Tumor",
splitPlotOnVar = "cnv_clusters" # Group by CNV clusters
)Custom Color Scheme
The heatmap uses a diverging color scale: - Blue: Deletions (negative CNV scores) - White: Neutral (no alteration) - Red: Amplifications (positive CNV scores)
# The default color scheme is optimized for CNV visualization
# Colors are scaled to the data range with balanced mappingHeatmap Components
A typical CNV heatmap includes:
┌─────────────────────────────────────────────────────────────┐
│ Chromosome Labels (1-22, X) │
├─────────────────────────────────────────────────────────────┤
│ Row │ │
│ Anno- │ CNV Score Matrix │
│ tations │ (cells × genomic windows) │
│ │ │
│ - Cell │ Blue = Deletion │
│ type │ White = Neutral │
│ - Clone │ Red = Amplification │
│ │ │
├─────────────────────────────────────────────────────────────┤
│ Color Legend │
└─────────────────────────────────────────────────────────────┘Phylogenetic Trees
Example Dendrogram
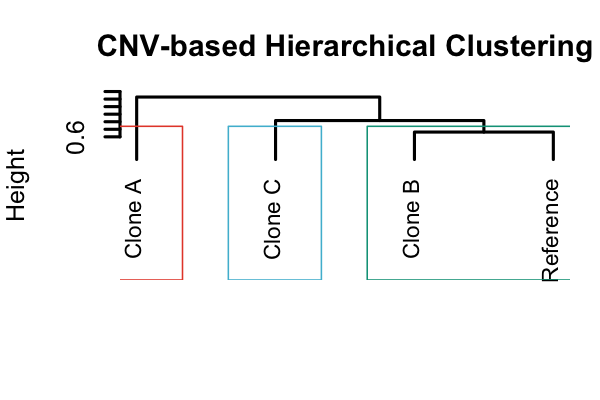
Hierarchical clustering dendrogram based on CNV profiles, showing the relationship between different clones.
Building CNV Trees
CNV profiles can be used to reconstruct clonal phylogenies:
# Build phylogenetic tree from CNV clusters
tree <- CNVTree(
seuratObj = result,
referenceVar = "cell_type",
tumorLabel = "Tumor",
cnv_thresh = 0.15
)Visualizing Trees
# Plot the phylogenetic tree
plotCNVTree(
tree = tree,
seuratObj = result,
referenceVar = "cell_type",
tumorLabel = "Tumor"
)Annotated Trees
Add CNV event annotations to tree branches:
# Annotate tree with CNV events
annotated_tree <- annotateCNVTree(
tree = tree,
seuratObj = result,
cnv_thresh = 0.15
)
# Plot with annotations
plotCNVTree(
tree = annotated_tree,
seuratObj = result,
referenceVar = "cell_type",
tumorLabel = "Tumor",
show_annotations = TRUE
)Chromosome Arm-Level Analysis
Example: CNV Fraction per Chromosome Arm
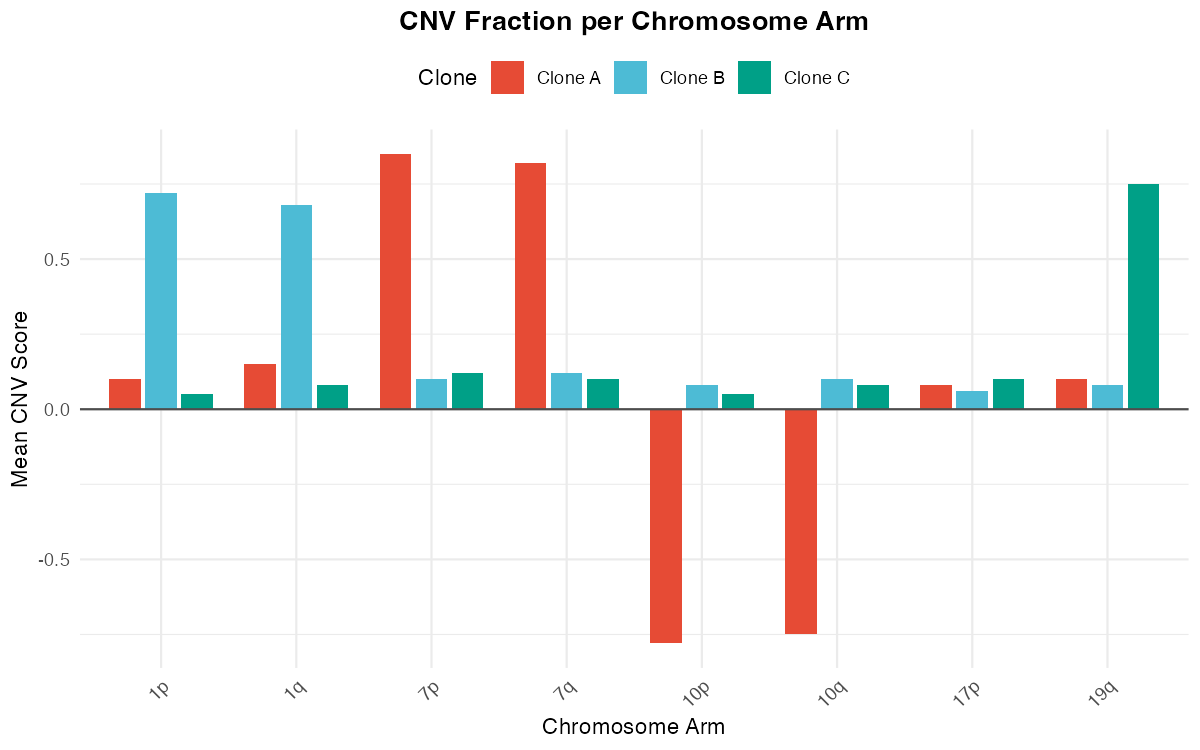
Bar plot showing mean CNV scores per chromosome arm for different clones.
Computing Arm-Level CNVs
# Calculate CNV fraction per chromosome arm
result <- CNVPerChromosomeArm(
seuratObj = result,
referenceVar = "cell_type",
tumorLabel = "Tumor"
)
# View arm-level data
head(result$cnv_per_arm)Visualizing Arm-Level CNVs
library(ggplot2)
# Extract arm-level data
arm_data <- result$cnv_per_arm
# Create bar plot
ggplot(arm_data, aes(x = arm, y = cnv_fraction, fill = cnv_type)) +
geom_bar(stat = "identity") +
facet_wrap(~cluster, ncol = 1) +
theme_minimal() +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
labs(
title = "CNV Fraction per Chromosome Arm",
x = "Chromosome Arm",
y = "Fraction of Cells with CNV"
)Spatial CNV Visualization
Visium Data
library(Seurat)
# Plot CNV clusters on tissue
SpatialDimPlot(
result,
group.by = "cnv_clusters",
pt.size.factor = 1.6
)Visium HD Data
# For Visium HD, use the specialized function
plotCNVResultsHD(
seuratObj = result,
referenceVar = "region",
tumorLabel = "Tumor"
)Combining Spatial and CNV Information
# Create a multi-panel figure
library(patchwork)
# Panel 1: Cell type annotation
p1 <- SpatialDimPlot(result, group.by = "cell_type")
# Panel 2: CNV clusters
p2 <- SpatialDimPlot(result, group.by = "cnv_clusters")
# Panel 3: CNV score for specific chromosome
p3 <- SpatialFeaturePlot(result, features = "chr7_cnv_score")
# Combine
p1 | p2 | p3Custom Visualizations
Creating Custom Plots
library(ggplot2)
library(tidyr)
# Example: Plot CNV profile for a single cell
cell_profile <- cnv_scores[, "cell_1"]
window_positions <- 1:length(cell_profile)
ggplot(data.frame(pos = window_positions, cnv = cell_profile)) +
geom_line(aes(x = pos, y = cnv), color = "steelblue") +
geom_hline(yintercept = 0, linetype = "dashed", color = "gray50") +
theme_minimal() +
labs(
title = "CNV Profile: Cell 1",
x = "Genomic Position (window)",
y = "CNV Score"
)Comparing Clusters
# Compare average CNV profiles between clusters
library(dplyr)
# Calculate mean CNV per cluster
cluster_means <- result@meta.data %>%
group_by(cnv_clusters) %>%
summarise(
mean_cnv_fraction = mean(cnv_fraction),
n_cells = n()
)
# Plot
ggplot(cluster_means, aes(x = cnv_clusters, y = mean_cnv_fraction, fill = cnv_clusters)) +
geom_bar(stat = "identity") +
theme_minimal() +
labs(
title = "Mean CNV Fraction by Cluster",
x = "CNV Cluster",
y = "Mean CNV Fraction"
)Publication-Ready Figures
High-Resolution Export
# Save heatmap as PDF
pdf("cnv_heatmap.pdf", width = 12, height = 8)
plotCNVResults(
seuratObj = result,
referenceVar = "cell_type",
tumorLabel = "Tumor"
)
dev.off()
# Save tree as PDF
pdf("cnv_tree.pdf", width = 8, height = 6)
plotCNVTree(
tree = tree,
seuratObj = result,
referenceVar = "cell_type",
tumorLabel = "Tumor"
)
dev.off()Figure Panel Assembly
library(patchwork)
library(ggplot2)
# Create individual panels
p_umap <- DimPlot(result, group.by = "cnv_clusters")
p_spatial <- SpatialDimPlot(result, group.by = "cnv_clusters")
# Assemble figure
figure <- (p_umap | p_spatial) /
plot_spacer() +
plot_layout(heights = c(2, 1))
# Save
ggsave("figure_panel.pdf", figure, width = 14, height = 10)Color Palettes
Default Palettes
fastCNV uses carefully selected color palettes:
# CNV score colors (diverging)
# Blue (-) → White (0) → Red (+)
# Cluster colors (qualitative)
# Uses paletteer for distinct, colorblind-friendly colors
library(paletteer)
cluster_colors <- paletteer_d("ggsci::default_nejm")Tips and Best Practices
1. Heatmap Ordering
- Order cells by cluster, then by similarity within cluster
- Use dendrograms to show hierarchical relationships
2. Color Scaling
- Center the color scale at zero
- Use symmetric limits for amplifications and deletions
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> loaded via a namespace (and not attached):
#> [1] digest_0.6.39 desc_1.4.3 R6_2.6.1 fastmap_1.2.0
#> [5] xfun_0.56 cachem_1.1.0 knitr_1.51 htmltools_0.5.9
#> [9] rmarkdown_2.30 lifecycle_1.0.5 cli_3.6.5 sass_0.4.10
#> [13] pkgdown_2.1.3 textshaping_1.0.4 jquerylib_0.1.4 systemfonts_1.3.1
#> [17] compiler_4.4.0 tools_4.4.0 ragg_1.5.0 bslib_0.9.0
#> [21] evaluate_1.0.5 yaml_2.3.12 otel_0.2.0 jsonlite_2.0.0
#> [25] rlang_1.1.7 fs_1.6.6 htmlwidgets_1.6.4