Introduction
iTALK provides three visualization functions for exploring cell-cell communication:
| Function | Type | Best For |
|---|---|---|
LRPlot() |
Circos plot | Detailed L-R pair visualization |
NetView() |
Network graph | Overall communication topology |
TimePlot() |
Time series | Dynamic changes across conditions |
This vignette demonstrates customization options for each visualization.
Setup
library(iTALK)
library(dplyr)
# Create example L-R pairs data
set.seed(42)
lr_data <- data.frame(
ligand = c("TGFB1", "VEGFA", "IL6", "CCL2", "CXCL12", "TNF", "IL1B", "PDGFA"),
receptor = c("TGFBR1", "KDR", "IL6R", "CCR2", "CXCR4", "TNFRSF1A", "IL1R1", "PDGFRA"),
cell_from = c("Macrophage", "Fibroblast", "T_cell", "Monocyte", "Stromal", "Macrophage", "Dendritic", "Fibroblast"),
cell_to = c("Fibroblast", "Endothelial", "B_cell", "T_cell", "T_cell", "Neutrophil", "T_cell", "Smooth_muscle"),
cell_from_mean_exprs = c(45, 32, 28, 55, 40, 38, 25, 42),
cell_to_mean_exprs = c(38, 45, 22, 35, 48, 30, 42, 35),
comm_type = c("growth_factor", "growth_factor", "cytokine", "chemokine", "chemokine", "cytokine", "cytokine", "growth_factor"),
stringsAsFactors = FALSE
)
# Define cell colors
cell_colors <- c(
"T_cell" = "#E41A1C",
"Macrophage" = "#377EB8",
"Fibroblast" = "#4DAF4A",
"B_cell" = "#984EA3",
"Monocyte" = "#FF7F00",
"Stromal" = "#A65628",
"Endothelial" = "#F781BF",
"Dendritic" = "#999999",
"Neutrophil" = "#66C2A5",
"Smooth_muscle" = "#FC8D62"
)
cat("Example data preview:\n")
#> Example data preview:
print(lr_data)
#> ligand receptor cell_from cell_to cell_from_mean_exprs
#> 1 TGFB1 TGFBR1 Macrophage Fibroblast 45
#> 2 VEGFA KDR Fibroblast Endothelial 32
#> 3 IL6 IL6R T_cell B_cell 28
#> 4 CCL2 CCR2 Monocyte T_cell 55
#> 5 CXCL12 CXCR4 Stromal T_cell 40
#> 6 TNF TNFRSF1A Macrophage Neutrophil 38
#> 7 IL1B IL1R1 Dendritic T_cell 25
#> 8 PDGFA PDGFRA Fibroblast Smooth_muscle 42
#> cell_to_mean_exprs comm_type
#> 1 38 growth_factor
#> 2 45 growth_factor
#> 3 22 cytokine
#> 4 35 chemokine
#> 5 48 chemokine
#> 6 30 cytokine
#> 7 42 cytokine
#> 8 35 growth_factorLRPlot: Circos Visualization
Basic Circos Plot
The LRPlot() function creates a circular visualization
where arrows represent ligand-receptor interactions between cell
types.
LRPlot(
data = lr_data,
datatype = "mean count",
cell_col = cell_colors,
transparency = 0.5,
link.arr.lwd = 2,
link.arr.type = "triangle"
)
Basic circos plot showing ligand-receptor interactions between cell types
Understanding the Circos Plot
The circos plot encodes multiple dimensions:
- Outer ring: Cell types (colored sectors)
- Inner ring: Gene names (ligands and receptors)
- Arrows: Communication direction (ligand → receptor)
- Arrow width: Expression level of ligand
- Arrow head width: Expression level of receptor
Customized Circos Plot
# Custom arrow properties based on expression
LRPlot(
data = lr_data,
datatype = "mean count",
cell_col = cell_colors,
transparency = 0.3,
link.arr.type = "big.arrow",
track.height_1 = circlize::uh(3, "mm"),
track.height_2 = circlize::uh(15, "mm"),
text.vjust = "0.5cm"
)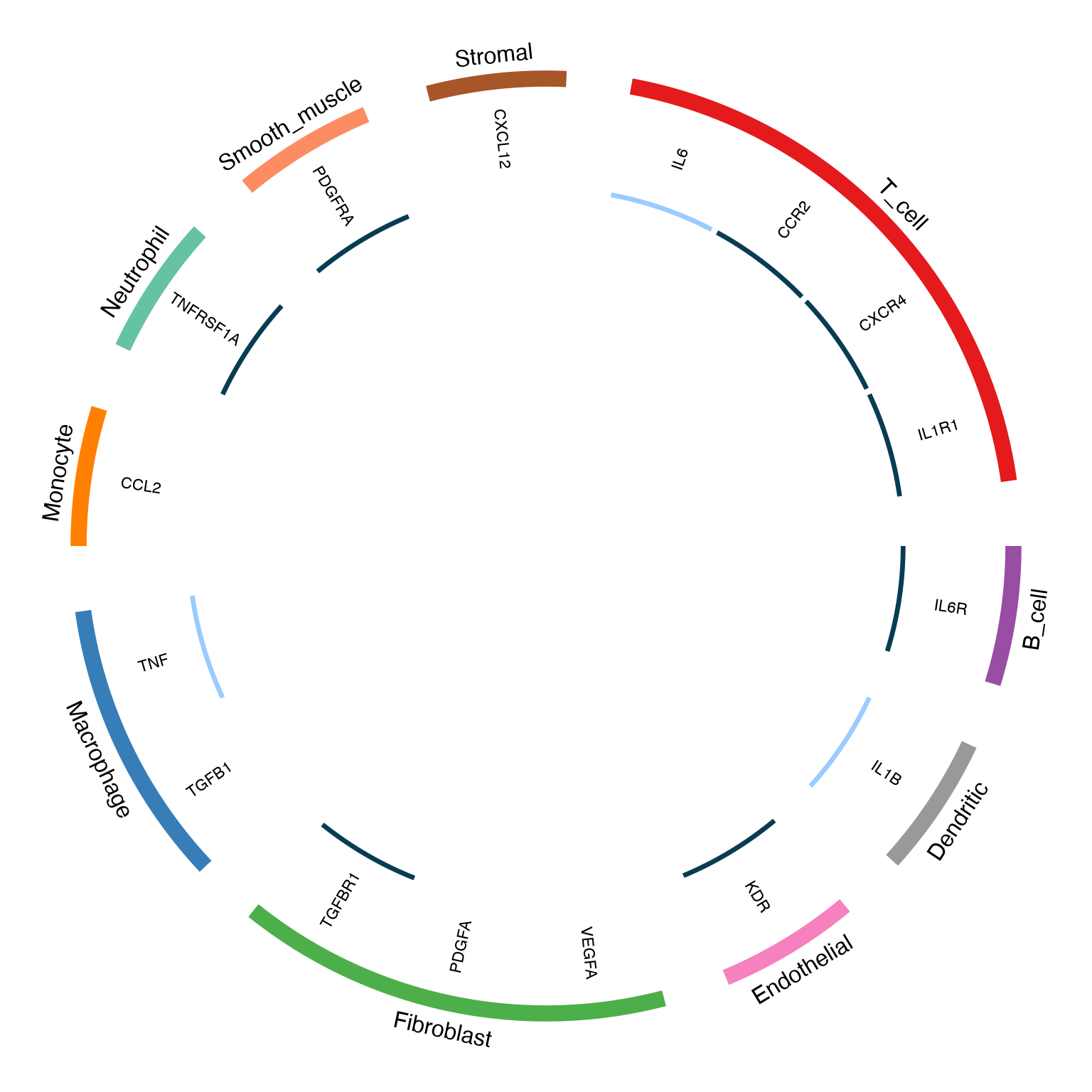
Customized circos plot with adjusted arrow properties
NetView: Network Visualization
Basic Network Plot
The NetView() function creates a network graph showing
overall communication patterns between cell types.
g <- NetView(
data = lr_data,
col = cell_colors,
vertex.size = 30,
vertex.label.cex = 0.8,
edge.max.width = 8,
edge.curved = 0.2,
arrow.width = 1.5,
margin = 0.2
)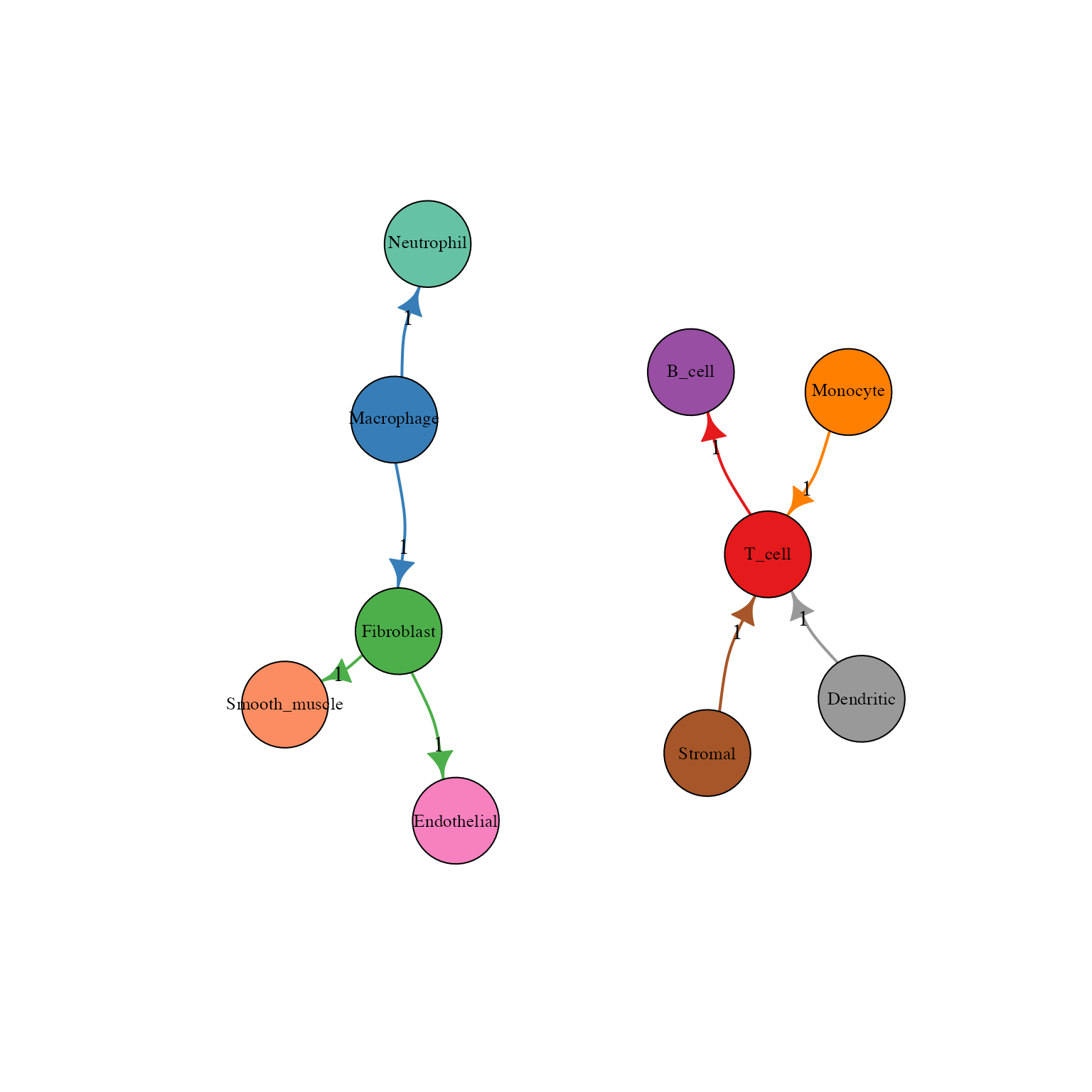
Network view of cell-cell communication
Understanding the Network
- Nodes: Cell types (size can represent number of interactions)
- Edges: Communication channels between cells
- Edge width: Number of L-R pairs
- Edge color: Source cell type color
- Arrows: Direction of communication
Customized Network with Labels
g <- NetView(
data = lr_data,
col = cell_colors,
vertex.size = 35,
vertex.label.cex = 0.9,
vertex.label.color = "white",
edge.max.width = 10,
edge.curved = 0.3,
arrow.width = 2,
edge.label.cex = 0.7,
edge.label.color = "darkgray",
label = TRUE,
margin = 0.15
)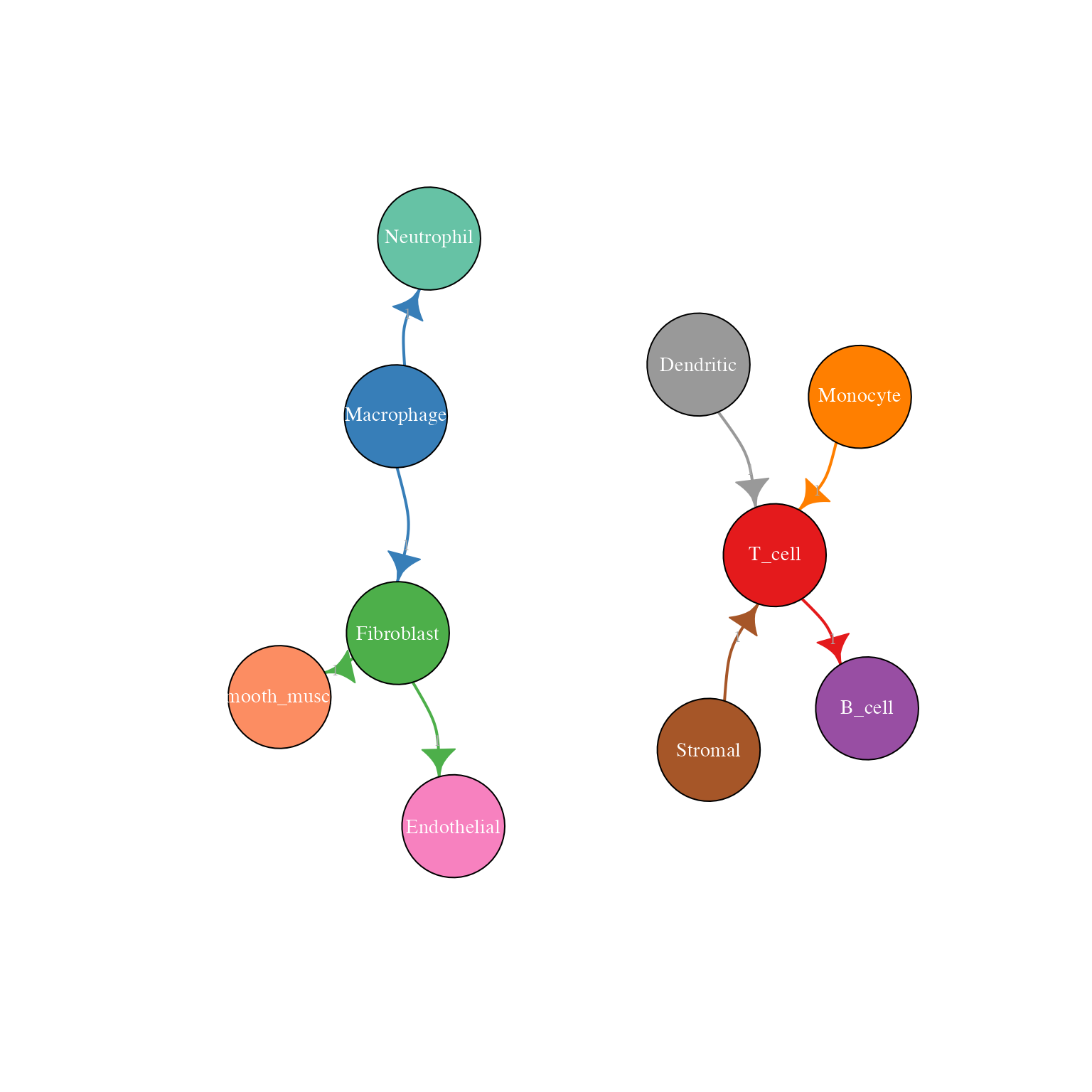
Network plot with edge labels showing interaction counts
DEG Data Visualization
For differential expression results, the visualization changes to show up/down regulation:
# Create DEG-style data
lr_deg <- data.frame(
ligand = c("TGFB1", "IL6", "CCL2", "TNF"),
receptor = c("TGFBR1", "IL6R", "CCR2", "TNFRSF1A"),
cell_from = c("Macrophage", "T_cell", "Monocyte", "Macrophage"),
cell_to = c("Fibroblast", "B_cell", "T_cell", "Neutrophil"),
cell_from_logFC = c(2.5, -1.8, 1.2, 3.0),
cell_to_logFC = c(1.8, 2.1, -0.8, 1.5),
comm_type = c("growth_factor", "cytokine", "chemokine", "cytokine"),
stringsAsFactors = FALSE
)
LRPlot(
data = lr_deg,
datatype = "DEG",
cell_col = cell_colors,
transparency = 0.4
)
Circos plot for DEG data showing up/down regulation
Arrow color coding for DEG:
| Ligand | Receptor | Arrow Color |
|---|---|---|
| ↑ Up | ↑ Up | Red (#d73027) |
| ↑ Up | ↓ Down | Yellow (#dfc27d) |
| ↓ Down | ↑ Up | Purple (#9933ff) |
| ↓ Down | ↓ Down | Cyan (#00ccff) |
Color Recommendations
Scientific Color Palettes
# Display recommended palettes
par(mfrow = c(1, 3), mar = c(1, 1, 2, 1))
# Option 1: Set1 from RColorBrewer
pal1 <- c("#E41A1C", "#377EB8", "#4DAF4A", "#984EA3", "#FF7F00", "#A65628")
barplot(rep(1, 6), col = pal1, border = NA, main = "Set1 Palette", axes = FALSE)
# Option 2: Paired
pal2 <- c("#A6CEE3", "#1F78B4", "#B2DF8A", "#33A02C", "#FB9A99", "#E31A1C")
barplot(rep(1, 6), col = pal2, border = NA, main = "Paired Palette", axes = FALSE)
# Option 3: Dark2
pal3 <- c("#1B9E77", "#D95F02", "#7570B3", "#E7298A", "#66A61E", "#E6AB02")
barplot(rep(1, 6), col = pal3, border = NA, main = "Dark2 Palette", axes = FALSE)
Publication-Ready Export
# PDF export for vector graphics
pdf("communication_circos.pdf", width = 10, height = 10)
LRPlot(lr_data, datatype = "mean count", cell_col = cell_colors)
dev.off()
# PNG export for high-resolution raster
png("communication_network.png", width = 3000, height = 3000, res = 300)
NetView(lr_data, col = cell_colors, vertex.size = 30)
dev.off()Tips and Best Practices
- Limit pairs for clarity: Show top 20-30 pairs in LRPlot for readability
- Consistent colors: Use the same cell colors across all plots
- Meaningful order: Sort data by expression/significance before plotting
- Vector formats: Use PDF for publication-quality figures
- Color blindness: Consider colorblind-friendly palettes
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] dplyr_1.1.4 iTALK_0.1.1
#>
#> loaded via a namespace (and not attached):
#> [1] RColorBrewer_1.1-3 RcppArmadillo_15.2.3-1
#> [3] jsonlite_2.0.0 shape_1.4.6.1
#> [5] magrittr_2.0.4 modeltools_0.2-24
#> [7] farver_2.1.2 rmarkdown_2.30
#> [9] GlobalOptions_0.1.3 fs_1.6.6
#> [11] zlibbioc_1.52.0 ragg_1.5.0
#> [13] vctrs_0.7.0 Cairo_1.7-0
#> [15] fastICA_1.2-7 scde_2.34.0
#> [17] progress_1.2.3 htmltools_0.5.9
#> [19] S4Arrays_1.6.0 curl_7.0.0
#> [21] SparseArray_1.6.2 sass_0.4.10
#> [23] bslib_0.9.0 HSMMSingleCell_1.26.0
#> [25] htmlwidgets_1.6.4 desc_1.4.3
#> [27] plyr_1.8.9 sandwich_3.1-1
#> [29] zoo_1.8-15 cachem_1.1.0
#> [31] igraph_2.2.1 lifecycle_1.0.5
#> [33] pkgconfig_2.0.3 Matrix_1.7-4
#> [35] R6_2.6.1 fastmap_1.2.0
#> [37] GenomeInfoDbData_1.2.13 MatrixGenerics_1.18.1
#> [39] digest_0.6.39 numDeriv_2016.8-1.1
#> [41] pcaMethods_1.98.0 colorspace_2.1-2
#> [43] miscTools_0.6-28 S4Vectors_0.44.0
#> [45] DESeq2_1.46.0 irlba_2.3.5.1
#> [47] textshaping_1.0.4 GenomicRanges_1.58.0
#> [49] extRemes_2.2-1 RMTstat_0.3.1
#> [51] mgcv_1.9-3 httr_1.4.7
#> [53] abind_1.4-8 compiler_4.4.0
#> [55] brew_1.0-10 S7_0.2.1
#> [57] BiocParallel_1.40.2 viridis_0.6.5
#> [59] MASS_7.3-65 quantreg_6.1
#> [61] MAST_1.32.0 DelayedArray_0.32.0
#> [63] rjson_0.2.23 tools_4.4.0
#> [65] otel_0.2.0 DDRTree_0.1.5
#> [67] nnet_7.3-20 glue_1.8.0
#> [69] nlme_3.1-168 grid_4.4.0
#> [71] Rtsne_0.17 cluster_2.1.8.1
#> [73] reshape2_1.4.5 generics_0.1.4
#> [75] gtable_0.3.6 monocle_2.34.0
#> [77] tidyr_1.3.2 hms_1.1.4
#> [79] data.table_1.18.0 flexmix_2.3-20
#> [81] XVector_0.46.0 BiocGenerics_0.52.0
#> [83] RANN_2.6.2 pillar_1.11.1
#> [85] stringr_1.6.0 Lmoments_1.3-2
#> [87] limma_3.62.2 circlize_0.4.17
#> [89] splines_4.4.0 Rook_1.2
#> [91] lattice_0.22-7 survival_3.8-3
#> [93] SparseM_1.84-2 gamlss.data_6.0-7
#> [95] tidyselect_1.2.1 SingleCellExperiment_1.28.1
#> [97] locfit_1.5-9.12 pbapply_1.7-4
#> [99] randomcoloR_1.1.0.1 knitr_1.51
#> [101] gridExtra_2.3 V8_8.0.1
#> [103] IRanges_2.40.1 edgeR_4.4.2
#> [105] SummarizedExperiment_1.36.0 stats4_4.4.0
#> [107] xfun_0.56 Biobase_2.66.0
#> [109] statmod_1.5.1 matrixStats_1.5.0
#> [111] pheatmap_1.0.13 leidenbase_0.1.36
#> [113] stringi_1.8.7 VGAM_1.1-14
#> [115] UCSC.utils_1.2.0 statnet.common_4.13.0
#> [117] yaml_2.3.12 evaluate_1.0.5
#> [119] codetools_0.2-20 bbmle_1.0.25.1
#> [121] DEsingle_1.26.0 tibble_3.3.1
#> [123] cli_3.6.5 systemfonts_1.3.1
#> [125] jquerylib_0.1.4 network_1.19.0
#> [127] dichromat_2.0-0.1 pscl_1.5.9
#> [129] Rcpp_1.1.1 GenomeInfoDb_1.42.3
#> [131] coda_0.19-4.1 bdsmatrix_1.3-7
#> [133] parallel_4.4.0 MatrixModels_0.5-4
#> [135] pkgdown_2.1.3 ggplot2_4.0.1
#> [137] prettyunits_1.2.0 gamlss.dist_6.1-1
#> [139] viridisLite_0.4.2 mvtnorm_1.3-3
#> [141] slam_0.1-55 scales_1.4.0
#> [143] gamlss_5.5-0 purrr_1.2.1
#> [145] crayon_1.5.3 combinat_0.0-8
#> [147] distillery_1.2-2 maxLik_1.5-2.1
#> [149] rlang_1.1.7