Algorithm and Statistical Methods
Zaoqu Liu
2026-01-23
Source:vignettes/algorithm.Rmd
algorithm.RmdOverview
This vignette describes the statistical methods and algorithms implemented in iTALK for intercellular communication analysis.
Ligand-Receptor Database
Database Structure
iTALK includes a curated database of 2,649 ligand-receptor pairs categorized into four functional groups:
Database Sources
The database integrates information from:
- Cytokines - Zhang et al., 2007; Cameron et al., 2000-2013
- Checkpoint molecules - Auslander et al., 2018
- Ligand-receptor pairs - Ramilowski et al., Nature Communications, 2015
# Database columns
cat("Database columns:", paste(colnames(db), collapse = ", "), "\n\n")
#> Database columns: Pair.Name, Ligand.ApprovedSymbol, Ligand.Name, Receptor.ApprovedSymbol, Receptor.Name, Classification
# Example entries
head(db[, c("Ligand.ApprovedSymbol", "Receptor.ApprovedSymbol", "Classification")])
#> Ligand.ApprovedSymbol Receptor.ApprovedSymbol Classification
#> 1 A2M LRP1 other
#> 2 AANAT MTNR1A other
#> 3 AANAT MTNR1B other
#> 4 ACE AGTR2 other
#> 5 ACE BDKRB2 other
#> 6 ADAM10 AXL otherStatistical Methods for Differential Expression
Method Comparison
iTALK supports seven statistical methods for identifying differentially expressed genes:
| Method | Model | Best For | Reference |
|---|---|---|---|
| Wilcox | Wilcoxon rank-sum | General purpose | - |
| DESeq2 | Negative binomial | Bulk RNA-seq | Love et al., 2014 |
| edgeR | Negative binomial + EB | Bulk RNA-seq | McCarthy et al., 2012 |
| MAST | Two-part hurdle | scRNA-seq | Finak et al., 2015 |
| monocle | Census-based | Trajectory analysis | Qiu et al., 2017 |
| SCDE | Bayesian | scRNA-seq | Kharchenko et al., 2014 |
| DEsingle | Zero-inflated NB | Dropout modeling | Miao et al., 2018 |
Wilcoxon Rank-Sum Test
The default method uses the Wilcoxon rank-sum test (Mann-Whitney U test):
Where is the rank of observation in the combined sample.
Log Fold Change Calculation:
For raw counts:
Where prevents division by zero.
For log-transformed data:
L-R Pair Detection Algorithm
Workflow
┌─────────────────┐
│ Gene List(s) │
│ (expr/DEG) │
└────────┬────────┘
│
▼
┌─────────────────┐
│ Species Check │──► Auto-conversion if needed
└────────┬────────┘
│
▼
┌─────────────────┐
│ Database Match │
│ Ligand ∩ Recep │
└────────┬────────┘
│
▼
┌─────────────────┐
│ Annotate Pairs │
│ + Cell Types │
└────────┬────────┘
│
▼
┌─────────────────┐
│ Filter & Output │
└─────────────────┘Matching Algorithm
For “mean count” datatype:
# Pseudocode
ligand_genes <- intersect(input_genes, database$Ligand)
receptor_genes <- intersect(input_genes, database$Receptor)
pairs <- database[database$Ligand %in% ligand_genes &
database$Receptor %in% receptor_genes, ]For “DEG” datatype, additional filtering removes pairs where both ligand and receptor are not differentially expressed (logFC = 0.0001 placeholder).
Multiple Testing Correction
All p-values are adjusted using the Benjamini-Hochberg procedure:
Where: - = i-th smallest p-value - = total number of tests - = adjusted p-value (FDR)
# Demonstration of BH correction
set.seed(42)
p_values <- c(runif(50, 0, 0.01), runif(50, 0.01, 1)) # Mix of significant and non-significant
q_values <- p.adjust(p_values, method = "BH")
# Visualize
par(mfrow = c(1, 2))
hist(p_values, breaks = 20, main = "Raw P-values", xlab = "P-value", col = "steelblue")
hist(q_values, breaks = 20, main = "BH-adjusted Q-values", xlab = "Q-value", col = "coral")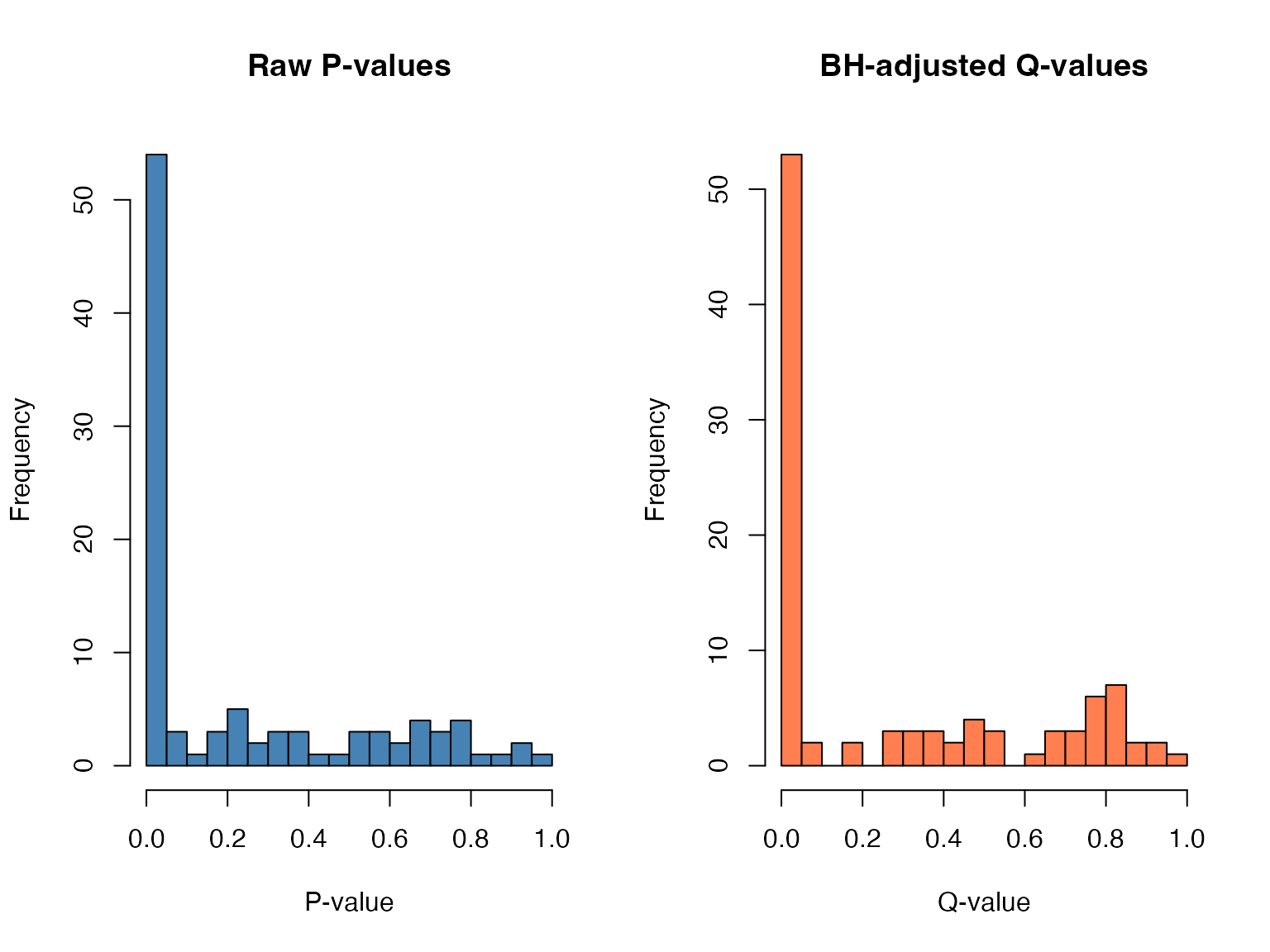
Communication Strength Metrics
Network Construction
Cell-cell communication networks are constructed using:
- Nodes = Cell types
- Edges = Number of L-R pairs between cell types
- Edge weights = Communication frequency or strength
library(igraph)
# Example network construction
edges <- data.frame(
from = c("T_cell", "T_cell", "Macrophage", "Dendritic"),
to = c("Macrophage", "B_cell", "T_cell", "T_cell"),
weight = c(15, 8, 12, 6)
)
g <- graph_from_data_frame(edges, directed = TRUE)
E(g)$width <- E(g)$weight / 3
plot(g,
vertex.size = 30,
vertex.color = "lightblue",
edge.arrow.size = 0.5,
main = "Cell Communication Network")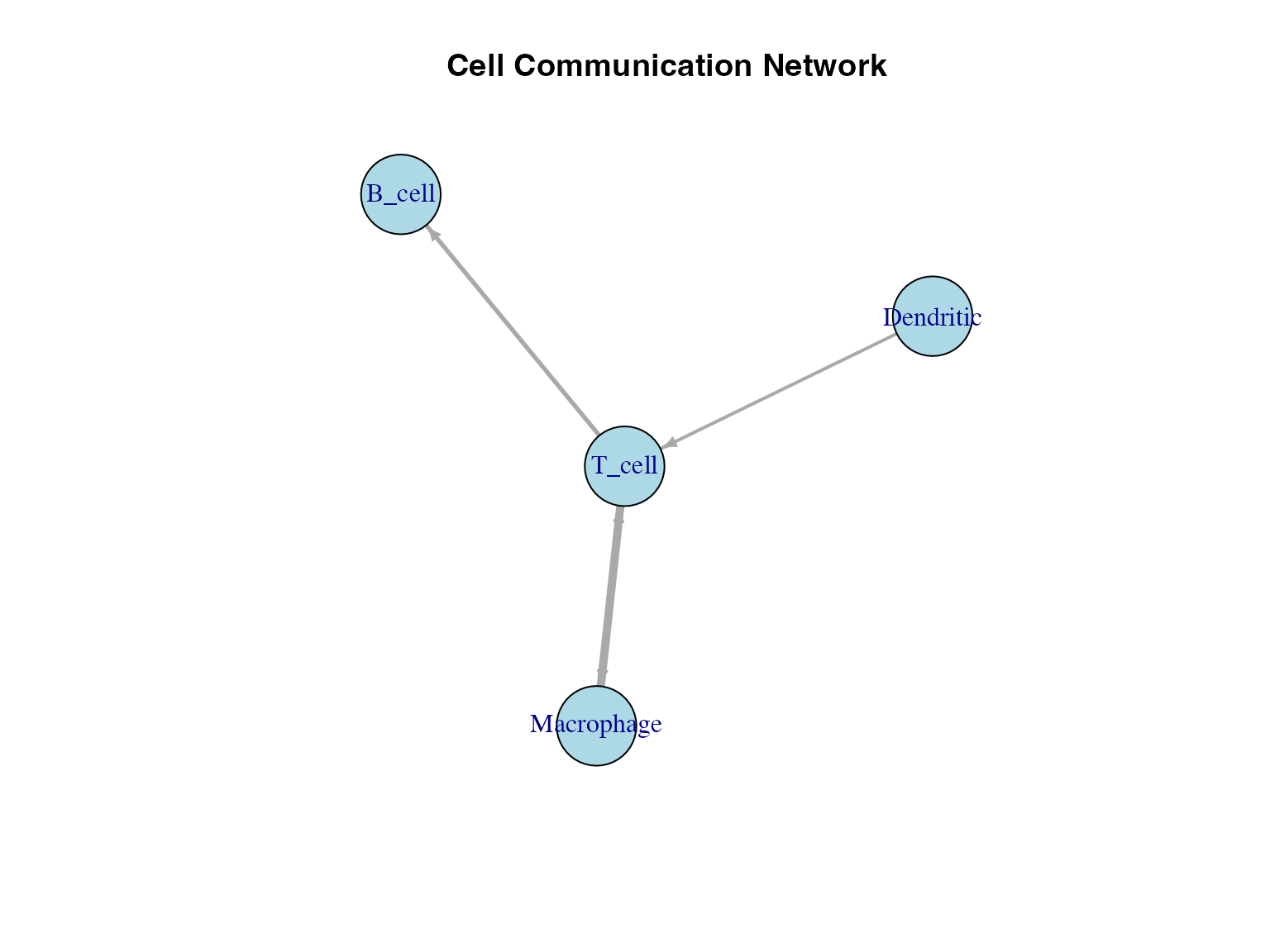
Computational Complexity
| Operation | Time Complexity | Space Complexity |
|---|---|---|
| rawParse | O(n × g) | O(c × g) |
| FindLR | O(g × d) | O(p) |
| DEG (Wilcox) | O(n × g) | O(g) |
| LRPlot | O(p) | O(p) |
| NetView | O(c²) | O(c²) |
Where: - n = number of cells - g = number of genes - c = number of cell types - d = database size - p = number of pairs
References
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014;15:550.
Finak G, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biology. 2015;16:278.
Wang Y, et al. iTALK: an R Package to Characterize and Illustrate Intercellular Communication. bioRxiv. 2019;507871.
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] igraph_2.2.1 iTALK_0.1.1
#>
#> loaded via a namespace (and not attached):
#> [1] RColorBrewer_1.1-3 RcppArmadillo_15.2.3-1
#> [3] jsonlite_2.0.0 shape_1.4.6.1
#> [5] magrittr_2.0.4 modeltools_0.2-24
#> [7] farver_2.1.2 rmarkdown_2.30
#> [9] GlobalOptions_0.1.3 fs_1.6.6
#> [11] zlibbioc_1.52.0 ragg_1.5.0
#> [13] vctrs_0.7.0 Cairo_1.7-0
#> [15] fastICA_1.2-7 scde_2.34.0
#> [17] progress_1.2.3 htmltools_0.5.9
#> [19] S4Arrays_1.6.0 curl_7.0.0
#> [21] SparseArray_1.6.2 sass_0.4.10
#> [23] bslib_0.9.0 HSMMSingleCell_1.26.0
#> [25] htmlwidgets_1.6.4 desc_1.4.3
#> [27] plyr_1.8.9 sandwich_3.1-1
#> [29] zoo_1.8-15 cachem_1.1.0
#> [31] lifecycle_1.0.5 pkgconfig_2.0.3
#> [33] Matrix_1.7-4 R6_2.6.1
#> [35] fastmap_1.2.0 GenomeInfoDbData_1.2.13
#> [37] MatrixGenerics_1.18.1 digest_0.6.39
#> [39] numDeriv_2016.8-1.1 pcaMethods_1.98.0
#> [41] colorspace_2.1-2 miscTools_0.6-28
#> [43] S4Vectors_0.44.0 DESeq2_1.46.0
#> [45] irlba_2.3.5.1 textshaping_1.0.4
#> [47] GenomicRanges_1.58.0 extRemes_2.2-1
#> [49] RMTstat_0.3.1 mgcv_1.9-3
#> [51] httr_1.4.7 abind_1.4-8
#> [53] compiler_4.4.0 brew_1.0-10
#> [55] S7_0.2.1 BiocParallel_1.40.2
#> [57] viridis_0.6.5 MASS_7.3-65
#> [59] quantreg_6.1 MAST_1.32.0
#> [61] DelayedArray_0.32.0 rjson_0.2.23
#> [63] tools_4.4.0 otel_0.2.0
#> [65] DDRTree_0.1.5 nnet_7.3-20
#> [67] glue_1.8.0 nlme_3.1-168
#> [69] grid_4.4.0 Rtsne_0.17
#> [71] cluster_2.1.8.1 reshape2_1.4.5
#> [73] generics_0.1.4 gtable_0.3.6
#> [75] monocle_2.34.0 tidyr_1.3.2
#> [77] hms_1.1.4 data.table_1.18.0
#> [79] flexmix_2.3-20 XVector_0.46.0
#> [81] BiocGenerics_0.52.0 RANN_2.6.2
#> [83] pillar_1.11.1 stringr_1.6.0
#> [85] Lmoments_1.3-2 limma_3.62.2
#> [87] circlize_0.4.17 splines_4.4.0
#> [89] dplyr_1.1.4 Rook_1.2
#> [91] lattice_0.22-7 survival_3.8-3
#> [93] SparseM_1.84-2 gamlss.data_6.0-7
#> [95] tidyselect_1.2.1 SingleCellExperiment_1.28.1
#> [97] locfit_1.5-9.12 pbapply_1.7-4
#> [99] randomcoloR_1.1.0.1 knitr_1.51
#> [101] gridExtra_2.3 V8_8.0.1
#> [103] IRanges_2.40.1 edgeR_4.4.2
#> [105] SummarizedExperiment_1.36.0 stats4_4.4.0
#> [107] xfun_0.56 Biobase_2.66.0
#> [109] statmod_1.5.1 matrixStats_1.5.0
#> [111] pheatmap_1.0.13 leidenbase_0.1.36
#> [113] stringi_1.8.7 VGAM_1.1-14
#> [115] UCSC.utils_1.2.0 statnet.common_4.13.0
#> [117] yaml_2.3.12 evaluate_1.0.5
#> [119] codetools_0.2-20 bbmle_1.0.25.1
#> [121] DEsingle_1.26.0 tibble_3.3.1
#> [123] cli_3.6.5 systemfonts_1.3.1
#> [125] jquerylib_0.1.4 network_1.19.0
#> [127] dichromat_2.0-0.1 pscl_1.5.9
#> [129] Rcpp_1.1.1 GenomeInfoDb_1.42.3
#> [131] coda_0.19-4.1 bdsmatrix_1.3-7
#> [133] parallel_4.4.0 MatrixModels_0.5-4
#> [135] pkgdown_2.1.3 ggplot2_4.0.1
#> [137] prettyunits_1.2.0 gamlss.dist_6.1-1
#> [139] viridisLite_0.4.2 mvtnorm_1.3-3
#> [141] slam_0.1-55 scales_1.4.0
#> [143] gamlss_5.5-0 purrr_1.2.1
#> [145] crayon_1.5.3 combinat_0.0-8
#> [147] distillery_1.2-2 maxLik_1.5-2.1
#> [149] rlang_1.1.7