Introduction
iTALK (intercellular communication analysis toolkit) is an R package designed to characterize and visualize cell-cell communication from transcriptomic data. This vignette provides a quick introduction to the core functionality.
Installation
# From R-Universe (recommended)
install.packages("iTALK", repos = "https://zaoqu-liu.r-universe.dev")
# From GitHub
remotes::install_github("Zaoqu-Liu/iTALK")Load Package and Create Example Data
library(iTALK)
library(dplyr)
# Access database to get real ligand-receptor pairs
db <- iTALK:::database
cyto_db <- db[db$Classification == "cytokine", ]
# Get some real ligands and receptors from the database
ligands <- head(unique(cyto_db$Ligand.ApprovedSymbol), 10)
receptors <- head(unique(cyto_db$Receptor.ApprovedSymbol), 10)
genes <- unique(c(ligands, receptors))
cat("Using genes from database:\n")
#> Using genes from database:
cat("Ligands:", paste(ligands, collapse = ", "), "\n")
#> Ligands: CCL11, CCL13, CCL14, CCL15, CCL16, CCL17, CCL18, CCL19, CCL1, CCL20
cat("Receptors:", paste(receptors, collapse = ", "), "\n\n")
#> Receptors: ACKR2, ACKR4, CCR2, CCR3, CCR5, CXCR3, CCR1, CCR9, CCR8, HRH4
# Create synthetic expression data
set.seed(42)
n_cells <- 200
cell_types <- c("T_cell", "B_cell", "Macrophage", "Dendritic", "NK_cell")
data <- data.frame(
cell_type = sample(cell_types, n_cells, replace = TRUE)
)
# Add gene expression
for (gene in genes) {
data[[gene]] <- rpois(n_cells, lambda = sample(5:25, 1))
}
cat("Data dimensions:", nrow(data), "cells x", ncol(data), "columns\n")
#> Data dimensions: 200 cells x 21 columns
cat("Cell types:", paste(unique(data$cell_type), collapse = ", "), "\n")
#> Cell types: T_cell, NK_cell, B_cell, Dendritic, Macrophage
head(data[, 1:6])
#> cell_type CCL11 CCL13 CCL14 CCL15 CCL16
#> 1 T_cell 11 22 21 7 13
#> 2 NK_cell 20 14 22 7 8
#> 3 T_cell 8 22 20 4 8
#> 4 T_cell 11 28 27 12 14
#> 5 B_cell 17 30 34 6 12
#> 6 Dendritic 12 19 18 7 7Workflow 1: Highly Expressed Gene Analysis
Step 1: Parse Expression Data
Identify the top expressed genes in each cell type:
# Get top 50% highly expressed genes per cell type
highly_expr <- rawParse(data, top_genes = 50, stats = "mean")
cat("\nTop expressed genes found:", nrow(highly_expr), "\n")
#>
#> Top expressed genes found: 50
cat("Cell types:", paste(unique(highly_expr$cell_type), collapse = ", "), "\n\n")
#> Cell types: T_cell, NK_cell, B_cell, Dendritic, Macrophage
head(highly_expr)
#> gene exprs cell_type
#> 1 CCL14 25.65306 T_cell
#> 2 CXCR3 24.55102 T_cell
#> 3 CCL13 24.24490 T_cell
#> 4 CCR8 23.89796 T_cell
#> 5 HRH4 19.02041 T_cell
#> 6 ACKR4 17.63265 T_cellStep 2: Find Ligand-Receptor Pairs
# Find ligand-receptor pairs from cytokine category
lr_pairs <- FindLR(
data_1 = highly_expr,
datatype = "mean count",
comm_type = "cytokine"
)
cat("Found", nrow(lr_pairs), "ligand-receptor pairs\n\n")
#> Found 243 ligand-receptor pairs
if (nrow(lr_pairs) > 0) {
head(lr_pairs)
}
#> ligand receptor cell_from_mean_exprs cell_from cell_to_mean_exprs cell_to
#> 1 CCL11 ACKR2 13.26531 T_cell 14.34694 T_cell
#> 2 CCL11 ACKR2 13.26531 T_cell 14.58974 NK_cell
#> 3 CCL11 ACKR2 13.26531 T_cell 15.08889 B_cell
#> 4 CCL11 ACKR2 13.26531 T_cell 13.60000 Dendritic
#> 5 CCL11 ACKR2 13.26531 T_cell 13.25926 Macrophage
#> 6 CCL11 ACKR2 12.30769 NK_cell 14.34694 T_cell
#> comm_type
#> 1 cytokine
#> 2 cytokine
#> 3 cytokine
#> 4 cytokine
#> 5 cytokine
#> 6 cytokineStep 3: Visualize with Circos Plot
# Define cell type colors
cell_col <- c(
"T_cell" = "#E41A1C",
"B_cell" = "#377EB8",
"Macrophage" = "#4DAF4A",
"Dendritic" = "#984EA3",
"NK_cell" = "#FF7F00"
)
# Create circos plot
if (nrow(lr_pairs) > 0) {
# Take top pairs for cleaner visualization
plot_data <- lr_pairs[1:min(20, nrow(lr_pairs)), ]
LRPlot(
data = plot_data,
datatype = "mean count",
cell_col = cell_col,
transparency = 0.5,
link.arr.type = "triangle"
)
}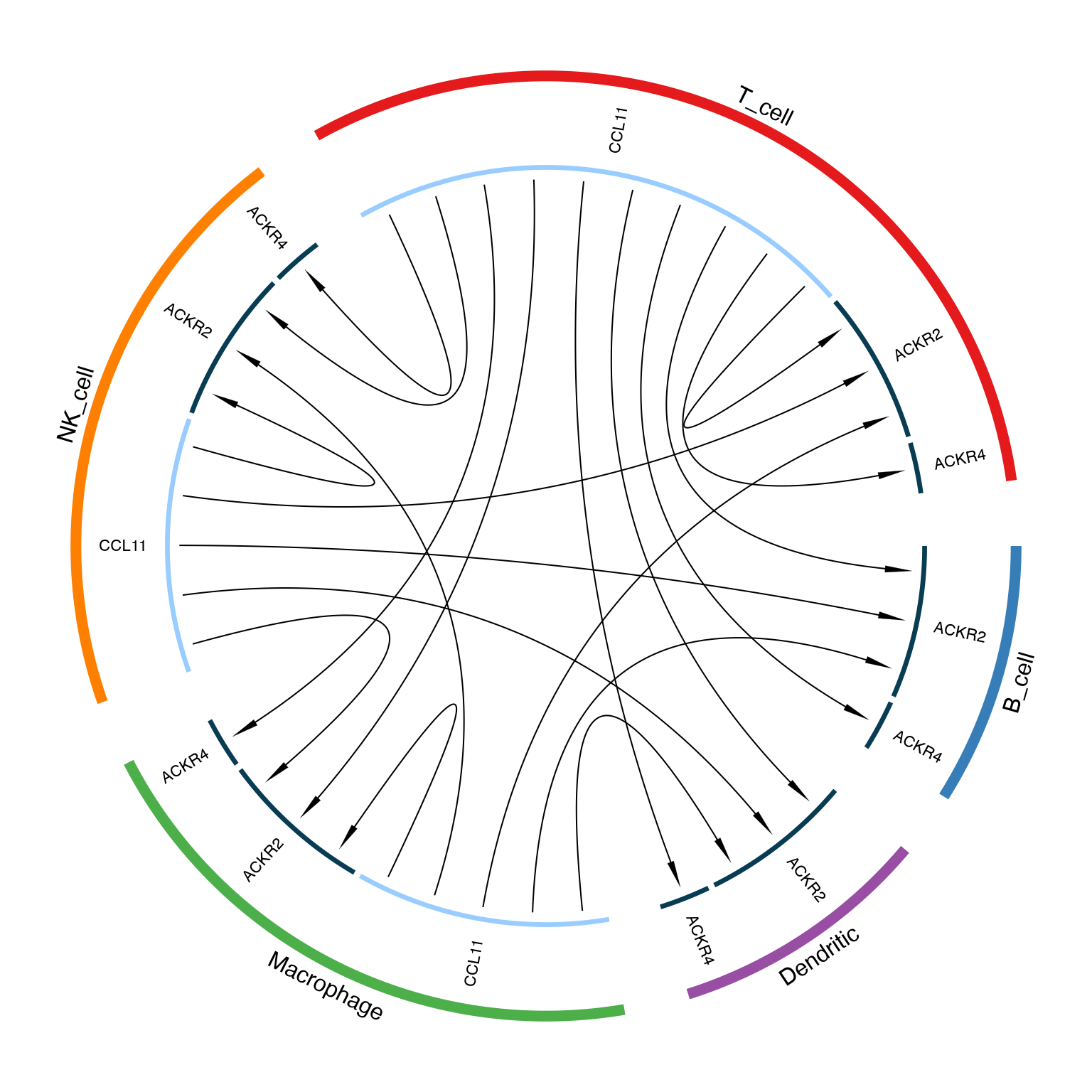
Circos plot showing ligand-receptor interactions between cell types
Workflow 2: Multi-Category Analysis
You can analyze different communication types separately:
# Communication categories in database
comm_types <- c("cytokine", "growth factor", "checkpoint", "other")
# Analyze each category
results_list <- list()
for (comm in comm_types) {
lr <- FindLR(highly_expr, datatype = "mean count", comm_type = comm)
if (nrow(lr) > 0) {
results_list[[comm]] <- lr
cat(comm, ":", nrow(lr), "pairs\n")
}
}
#> cytokine : 243 pairsWorkflow 3: Using the Ligand-Receptor Database
iTALK includes a curated database of ligand-receptor pairs:
# Access the built-in database
data(database)
cat("Database contains", nrow(database), "ligand-receptor pairs\n\n")
#> Database contains 2649 ligand-receptor pairs
cat("Categories:", paste(unique(database$Classification), collapse = ", "), "\n\n")
#> Categories: other, checkpoint, cytokine, growth factor
# Preview
head(database[, c("Pair.Name", "Ligand.ApprovedSymbol", "Receptor.ApprovedSymbol", "Classification")])
#> Pair.Name Ligand.ApprovedSymbol Receptor.ApprovedSymbol Classification
#> 1 A2M_LRP1 A2M LRP1 other
#> 2 AANAT_MTNR1A AANAT MTNR1A other
#> 3 AANAT_MTNR1B AANAT MTNR1B other
#> 4 ACE_AGTR2 ACE AGTR2 other
#> 5 ACE_BDKRB2 ACE BDKRB2 other
#> 6 ADAM10_AXL ADAM10 AXL otherSummary
This quick start covered:
- Data preparation - Expression matrix with cell type annotations
- Gene parsing - Identifying highly expressed genes per cell type
- L-R detection - Finding ligand-receptor pairs from the database
- Visualization - Circos and network plots
For more advanced usage, see:
-
vignette("algorithm")- Statistical methods and database -
vignette("species-conversion")- Cross-species analysis -
vignette("visualization")- Comprehensive visualization guide
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] dplyr_1.1.4 iTALK_0.1.1
#>
#> loaded via a namespace (and not attached):
#> [1] RColorBrewer_1.1-3 RcppArmadillo_15.2.3-1
#> [3] jsonlite_2.0.0 shape_1.4.6.1
#> [5] magrittr_2.0.4 modeltools_0.2-24
#> [7] farver_2.1.2 rmarkdown_2.30
#> [9] GlobalOptions_0.1.3 fs_1.6.6
#> [11] zlibbioc_1.52.0 ragg_1.5.0
#> [13] vctrs_0.7.0 Cairo_1.7-0
#> [15] fastICA_1.2-7 scde_2.34.0
#> [17] progress_1.2.3 htmltools_0.5.9
#> [19] S4Arrays_1.6.0 curl_7.0.0
#> [21] SparseArray_1.6.2 sass_0.4.10
#> [23] bslib_0.9.0 HSMMSingleCell_1.26.0
#> [25] htmlwidgets_1.6.4 desc_1.4.3
#> [27] plyr_1.8.9 sandwich_3.1-1
#> [29] zoo_1.8-15 cachem_1.1.0
#> [31] igraph_2.2.1 lifecycle_1.0.5
#> [33] pkgconfig_2.0.3 Matrix_1.7-4
#> [35] R6_2.6.1 fastmap_1.2.0
#> [37] GenomeInfoDbData_1.2.13 MatrixGenerics_1.18.1
#> [39] digest_0.6.39 numDeriv_2016.8-1.1
#> [41] pcaMethods_1.98.0 colorspace_2.1-2
#> [43] miscTools_0.6-28 S4Vectors_0.44.0
#> [45] DESeq2_1.46.0 irlba_2.3.5.1
#> [47] textshaping_1.0.4 GenomicRanges_1.58.0
#> [49] extRemes_2.2-1 RMTstat_0.3.1
#> [51] mgcv_1.9-3 httr_1.4.7
#> [53] abind_1.4-8 compiler_4.4.0
#> [55] withr_3.0.2 brew_1.0-10
#> [57] S7_0.2.1 BiocParallel_1.40.2
#> [59] viridis_0.6.5 MASS_7.3-65
#> [61] quantreg_6.1 MAST_1.32.0
#> [63] DelayedArray_0.32.0 rjson_0.2.23
#> [65] tools_4.4.0 otel_0.2.0
#> [67] DDRTree_0.1.5 nnet_7.3-20
#> [69] glue_1.8.0 nlme_3.1-168
#> [71] grid_4.4.0 Rtsne_0.17
#> [73] cluster_2.1.8.1 reshape2_1.4.5
#> [75] generics_0.1.4 gtable_0.3.6
#> [77] monocle_2.34.0 tidyr_1.3.2
#> [79] hms_1.1.4 data.table_1.18.0
#> [81] flexmix_2.3-20 XVector_0.46.0
#> [83] BiocGenerics_0.52.0 RANN_2.6.2
#> [85] pillar_1.11.1 stringr_1.6.0
#> [87] Lmoments_1.3-2 limma_3.62.2
#> [89] circlize_0.4.17 splines_4.4.0
#> [91] Rook_1.2 lattice_0.22-7
#> [93] survival_3.8-3 SparseM_1.84-2
#> [95] gamlss.data_6.0-7 tidyselect_1.2.1
#> [97] SingleCellExperiment_1.28.1 locfit_1.5-9.12
#> [99] pbapply_1.7-4 randomcoloR_1.1.0.1
#> [101] knitr_1.51 gridExtra_2.3
#> [103] V8_8.0.1 IRanges_2.40.1
#> [105] edgeR_4.4.2 SummarizedExperiment_1.36.0
#> [107] stats4_4.4.0 xfun_0.56
#> [109] Biobase_2.66.0 statmod_1.5.1
#> [111] matrixStats_1.5.0 pheatmap_1.0.13
#> [113] leidenbase_0.1.36 stringi_1.8.7
#> [115] VGAM_1.1-14 UCSC.utils_1.2.0
#> [117] statnet.common_4.13.0 yaml_2.3.12
#> [119] evaluate_1.0.5 codetools_0.2-20
#> [121] bbmle_1.0.25.1 DEsingle_1.26.0
#> [123] tibble_3.3.1 cli_3.6.5
#> [125] systemfonts_1.3.1 jquerylib_0.1.4
#> [127] network_1.19.0 dichromat_2.0-0.1
#> [129] pscl_1.5.9 Rcpp_1.1.1
#> [131] GenomeInfoDb_1.42.3 coda_0.19-4.1
#> [133] bdsmatrix_1.3-7 parallel_4.4.0
#> [135] MatrixModels_0.5-4 pkgdown_2.1.3
#> [137] ggplot2_4.0.1 prettyunits_1.2.0
#> [139] gamlss.dist_6.1-1 viridisLite_0.4.2
#> [141] mvtnorm_1.3-3 slam_0.1-55
#> [143] scales_1.4.0 gamlss_5.5-0
#> [145] purrr_1.2.1 crayon_1.5.3
#> [147] combinat_0.0-8 distillery_1.2-2
#> [149] maxLik_1.5-2.1 rlang_1.1.7