Algorithm Principles
Zaoqu Liu, Robert K. Suter, Nagi G. Ayad
2026-02-03
Source:vignettes/algorithm-principles.Rmd
algorithm-principles.RmdOverview
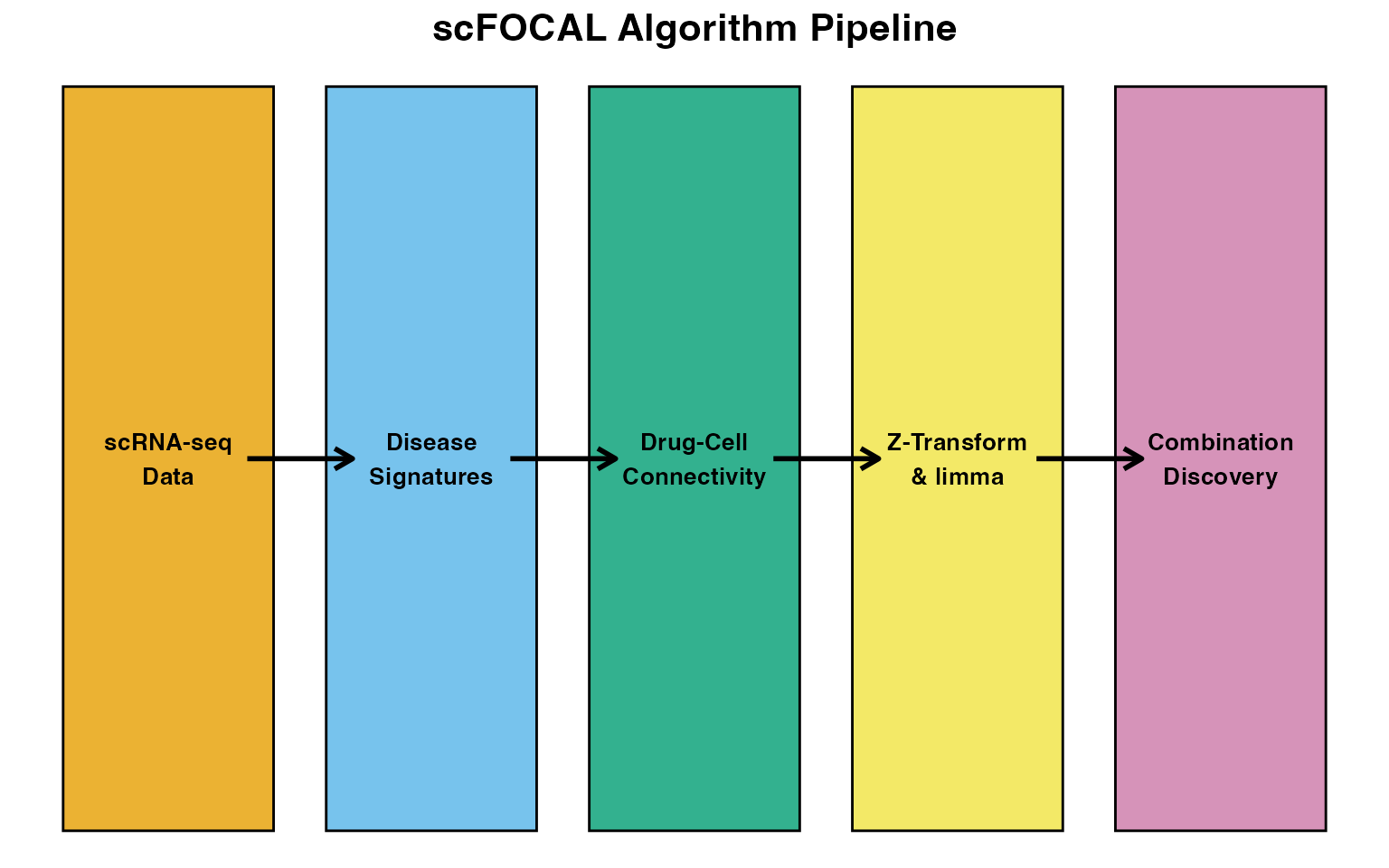
scFOCAL implements a multi-step computational framework that integrates single-cell transcriptomics with pharmacological perturbation data. This vignette details the mathematical principles underlying each algorithm.
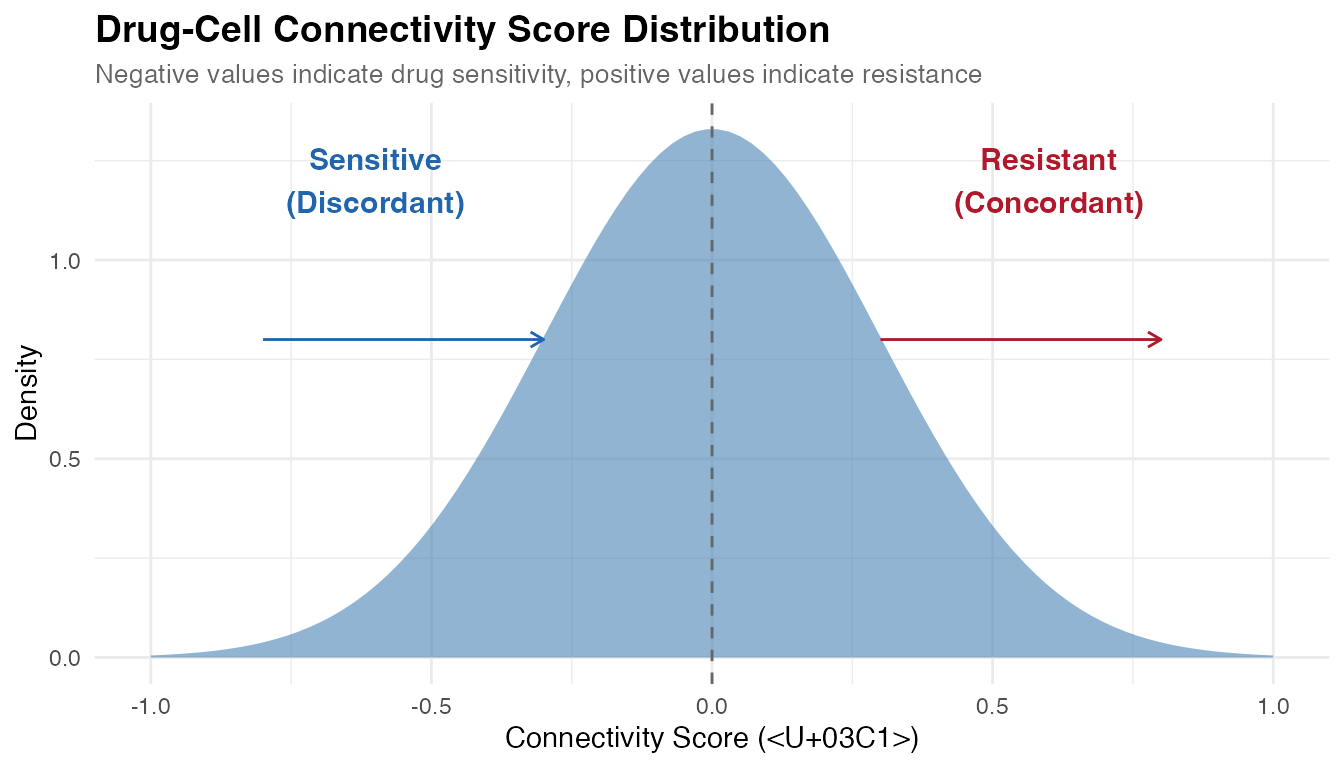
1. Drug-Cell Connectivity Score
Concept
The core innovation of scFOCAL is the Drug-Cell Connectivity Score, which quantifies the transcriptional relationship between individual cells and drug perturbation signatures.
2. Fisher’s Z-Transformation
Purpose
To enable statistical comparison of correlation coefficients across conditions, we apply Fisher’s Z-transformation:
Properties
# Demonstrate Fisher Z transformation
rho <- seq(-0.95, 0.95, by = 0.01)
z <- 0.5 * log((1 + rho) / (1 - rho))
df <- data.frame(rho = rho, z = z)
ggplot(df, aes(x = rho, y = z)) +
geom_line(color = "#0072B2", linewidth = 1.2) +
geom_hline(yintercept = 0, linetype = "dashed", color = "gray50") +
geom_vline(xintercept = 0, linetype = "dashed", color = "gray50") +
labs(
title = "Fisher's Z-Transformation",
subtitle = expression(Z == frac(1,2) * ln * bgroup("(", frac(1+rho, 1-rho), ")")),
x = expression("Correlation coefficient (" * rho * ")"),
y = "Fisher's Z"
) +
theme_minimal() +
theme(
plot.title = element_text(face = "bold", size = 14),
plot.subtitle = element_text(size = 12)
)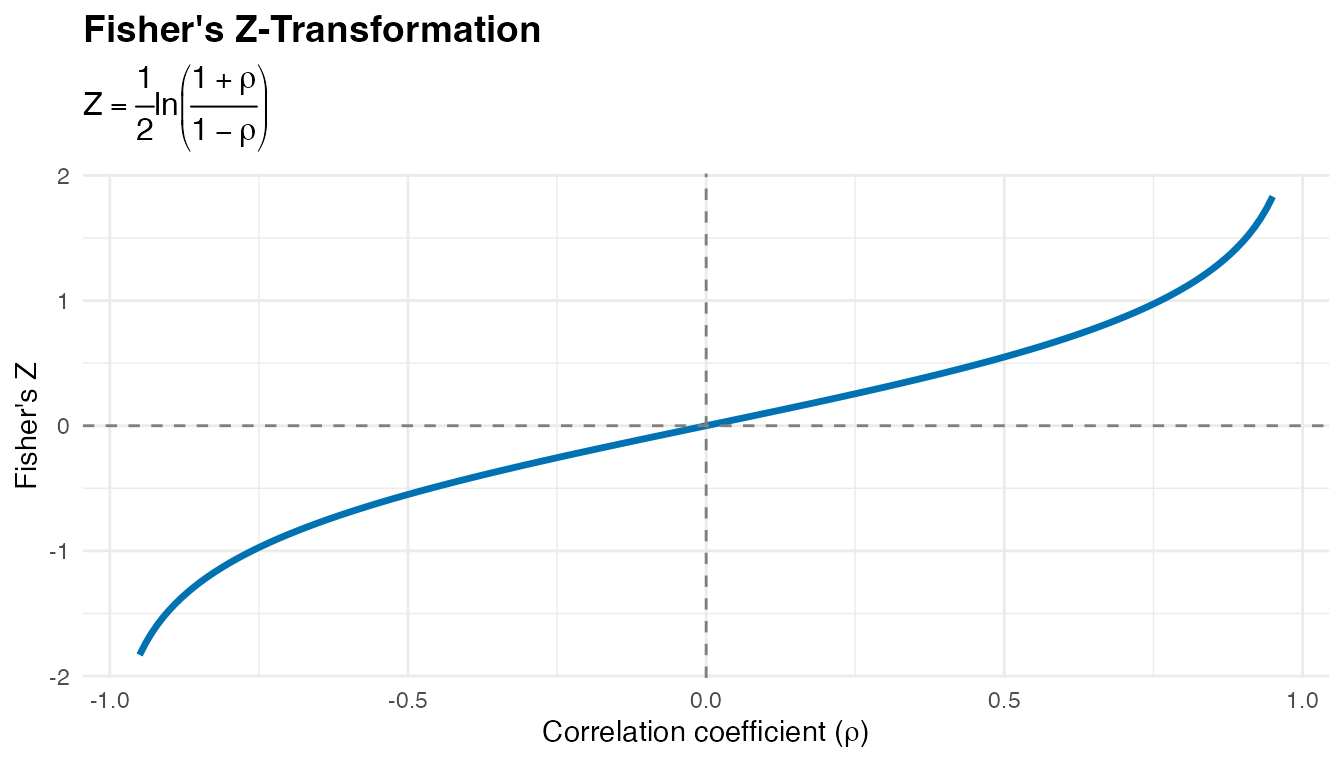
Key advantages:
- Normalizes the sampling distribution - Z-values follow an approximately normal distribution
- Stabilizes variance - Variance becomes approximately constant across different correlation values
- Enables parametric testing - Allows use of standard statistical tests (t-tests, ANOVA)
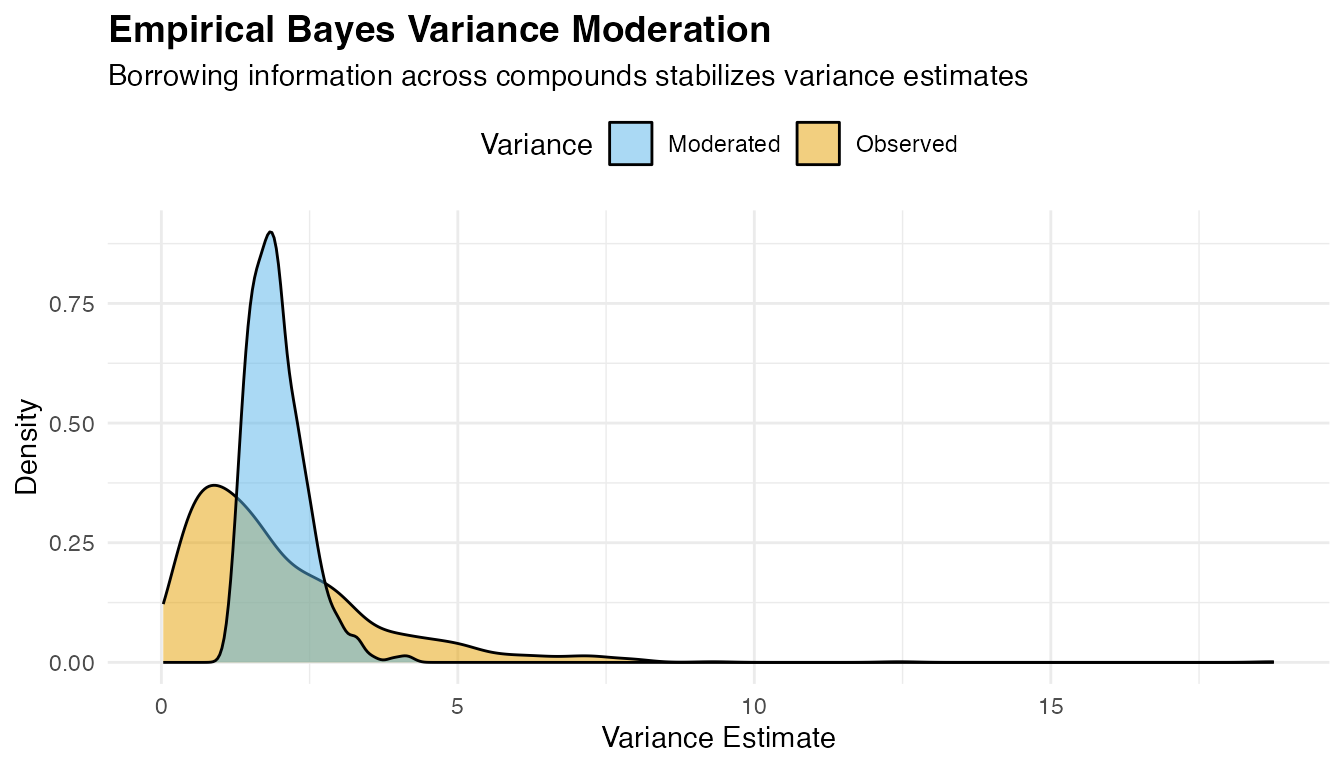
3. Differential Connectivity Analysis
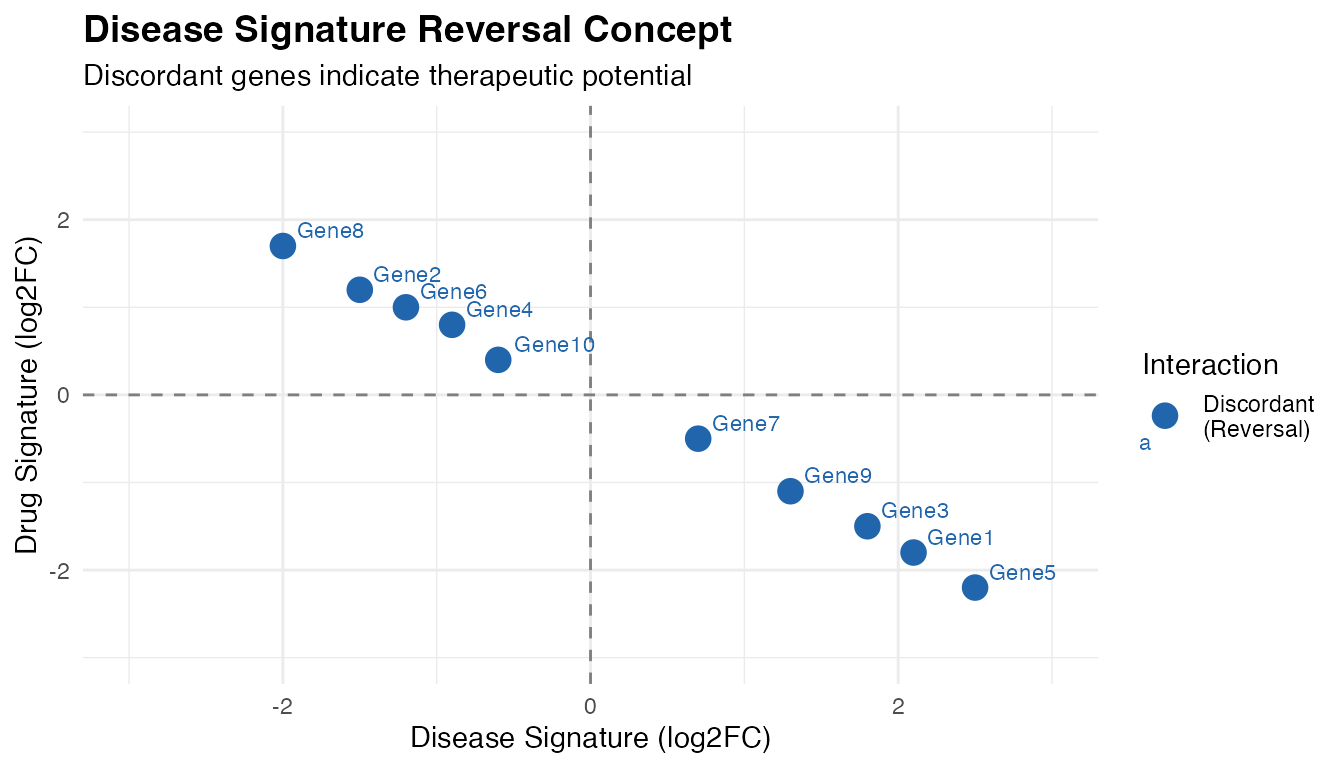
4. Disease Signature Reversal Score
5. MAST Differential Expression
6. Computational Complexity
Time Complexity Analysis
| Operation | Complexity | Typical Runtime |
|---|---|---|
| Drug-Cell Connectivity | O(n × m × g) | ~20 min for 10K cells |
| MAST Differential Expression | O(n × g) | ~5 min per comparison |
| Reversal Scoring | O(d × g) | < 1 min |
| Differential Connectivity | O(n × d) | ~2 min |
Where: n = cells, m = compounds (1679), g = genes (~1000), d = drug signatures
Summary
scFOCAL’s algorithmic framework provides:
- Robust statistical inference through Fisher Z-transformation and empirical Bayes
- Biological interpretability via connectivity and reversal scores
- Scalability to large single-cell datasets
- Integration of orthogonal pharmacological and transcriptomic data
References
- Subramanian A, et al. A Next Generation Connectivity Map. Cell (2017)
- Finak G, et al. MAST: a flexible statistical framework. Genome Biology (2015)
- Ritchie ME, et al. limma powers differential expression. Nucleic Acids Res (2015)
- Fisher RA. On the probable error of a coefficient of correlation. Metron (1921)
Session Info
## R version 4.4.0 (2024-04-24)
## Platform: aarch64-apple-darwin20
## Running under: macOS 15.6.1
##
## Matrix products: default
## BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
##
## locale:
## [1] C
##
## time zone: Asia/Shanghai
## tzcode source: internal
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] dplyr_1.1.4 ggplot2_4.0.1
##
## loaded via a namespace (and not attached):
## [1] gtable_0.3.6 jsonlite_2.0.0 compiler_4.4.0 tidyselect_1.2.1
## [5] dichromat_2.0-0.1 jquerylib_0.1.4 systemfonts_1.3.1 scales_1.4.0
## [9] textshaping_1.0.4 yaml_2.3.12 fastmap_1.2.0 R6_2.6.1
## [13] labeling_0.4.3 generics_0.1.4 knitr_1.51 htmlwidgets_1.6.4
## [17] tibble_3.3.1 desc_1.4.3 bslib_0.9.0 pillar_1.11.1
## [21] RColorBrewer_1.1-3 rlang_1.1.7 cachem_1.1.0 xfun_0.56
## [25] fs_1.6.6 sass_0.4.10 S7_0.2.1 otel_0.2.0
## [29] cli_3.6.5 pkgdown_2.2.0 withr_3.0.2 magrittr_2.0.4
## [33] digest_0.6.39 grid_4.4.0 lifecycle_1.0.5 vctrs_0.7.1
## [37] evaluate_1.0.5 glue_1.8.0 farver_2.1.2 ragg_1.5.0
## [41] rmarkdown_2.30 tools_4.4.0 pkgconfig_2.0.3 htmltools_0.5.9