Visualization Gallery
Zaoqu Liu, Robert K. Suter, Nagi G. Ayad
2026-02-03
Source:vignettes/visualization-gallery.Rmd
visualization-gallery.RmdIntroduction
scFOCAL generates a variety of publication-ready visualizations. This gallery showcases the main output types with customization examples.
1. Dimensional Reduction Plots
UMAP with Cell Type Annotations
set.seed(42)
n_cells <- 1500
# Generate mock UMAP data
umap_data <- data.frame(
UMAP1 = c(rnorm(300, -3, 0.8), rnorm(300, 0, 0.9), rnorm(300, 3, 0.8),
rnorm(300, -1.5, 0.7), rnorm(300, 1.5, 0.7)),
UMAP2 = c(rnorm(300, 2, 0.7), rnorm(300, 3, 0.8), rnorm(300, 1, 0.7),
rnorm(300, -2, 0.8), rnorm(300, -1, 0.7)),
CellType = factor(rep(c("Mesenchymal", "Astrocyte-like", "Neural Progenitor",
"Immune", "Stromal"), each = 300))
)
ggplot(umap_data, aes(x = UMAP1, y = UMAP2, color = CellType)) +
geom_point(size = 0.8, alpha = 0.7) +
scale_color_brewer(palette = "Set2", name = "Cell Type") +
labs(
title = "Single-Cell UMAP Projection",
subtitle = "Cell type annotations",
x = "UMAP 1",
y = "UMAP 2"
) +
theme_minimal() +
theme(
legend.position = "right",
plot.title = element_text(face = "bold", size = 14)
) +
guides(color = guide_legend(override.aes = list(size = 3)))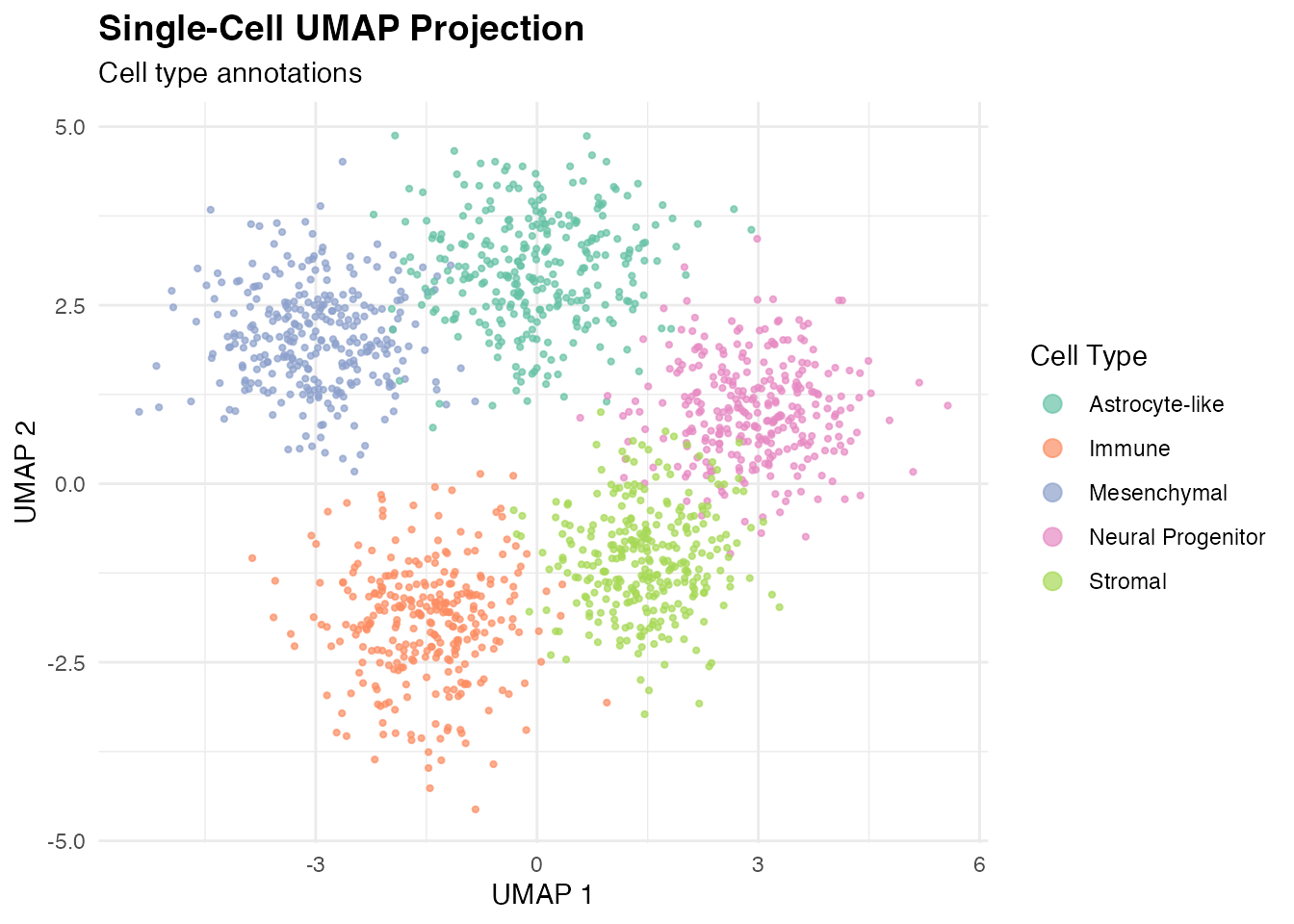
UMAP with Drug Connectivity
umap_data$Connectivity <- c(
rnorm(300, -0.15, 0.1), # MES - sensitive
rnorm(300, 0.1, 0.12), # AC - resistant
rnorm(300, 0.05, 0.1), # NPC
rnorm(300, 0, 0.08), # Immune
rnorm(300, 0, 0.08) # Stromal
)
ggplot(umap_data, aes(x = UMAP1, y = UMAP2, color = Connectivity)) +
geom_point(size = 0.8, alpha = 0.7) +
scale_color_gradient2(
low = "#2166AC",
mid = "white",
high = "#B2182B",
midpoint = 0,
name = "Drug\nConnectivity"
) +
labs(
title = "Drug-Cell Connectivity Score",
subtitle = "Temozolomide response prediction",
x = "UMAP 1",
y = "UMAP 2"
) +
theme_minimal() +
theme(plot.title = element_text(face = "bold", size = 14))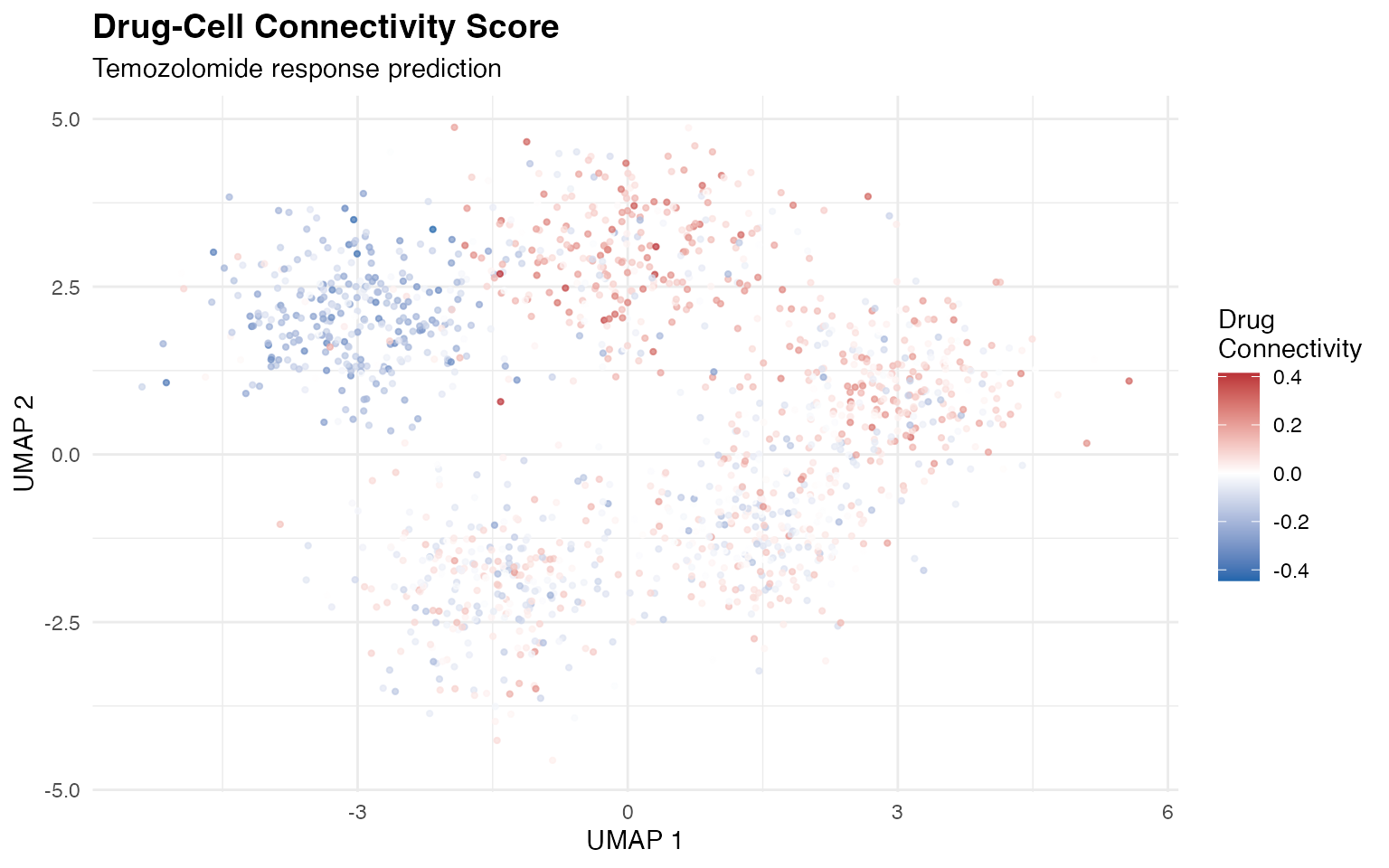
2. Violin Plots
Drug Connectivity by Cell Type
tumor_data <- umap_data[umap_data$CellType %in% c("Mesenchymal", "Astrocyte-like", "Neural Progenitor"),]
ggplot(tumor_data, aes(x = CellType, y = Connectivity, fill = CellType)) +
geom_violin(alpha = 0.7, scale = "width") +
geom_boxplot(width = 0.15, fill = "white", outlier.size = 0.5) +
geom_hline(yintercept = 0, linetype = "dashed", color = "gray40") +
scale_fill_brewer(palette = "Set2") +
labs(
title = "Drug Sensitivity by Tumor State",
subtitle = "Violin plot with embedded boxplot",
x = "",
y = "Drug-Cell Connectivity"
) +
theme_minimal() +
theme(
legend.position = "none",
axis.text.x = element_text(angle = 30, hjust = 1),
plot.title = element_text(face = "bold", size = 14)
) +
annotate("text", x = 3.4, y = -0.3, label = "Sensitive",
color = "#2166AC", fontface = "italic", size = 3.5) +
annotate("text", x = 3.4, y = 0.3, label = "Resistant",
color = "#B2182B", fontface = "italic", size = 3.5)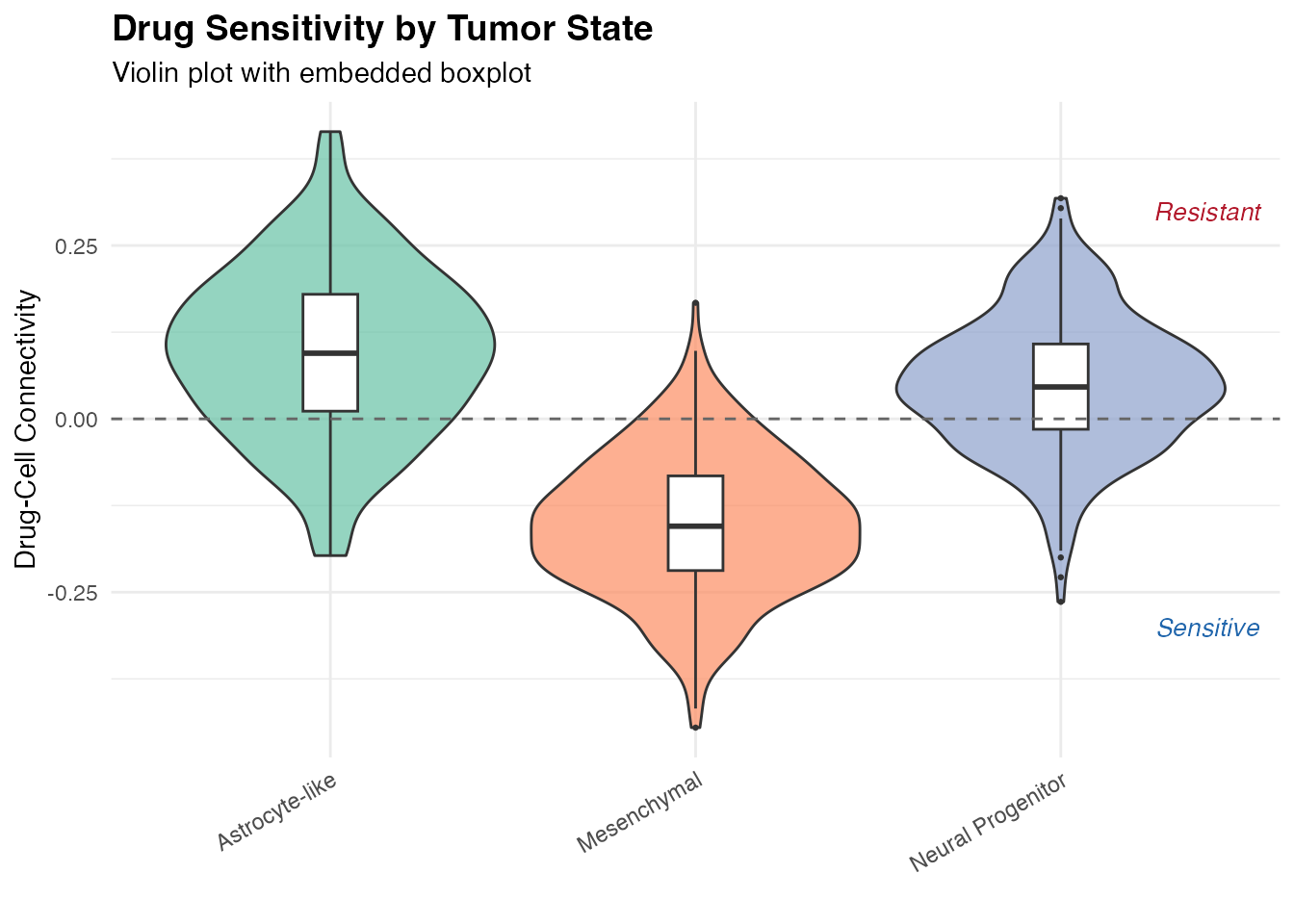
3. Heatmaps
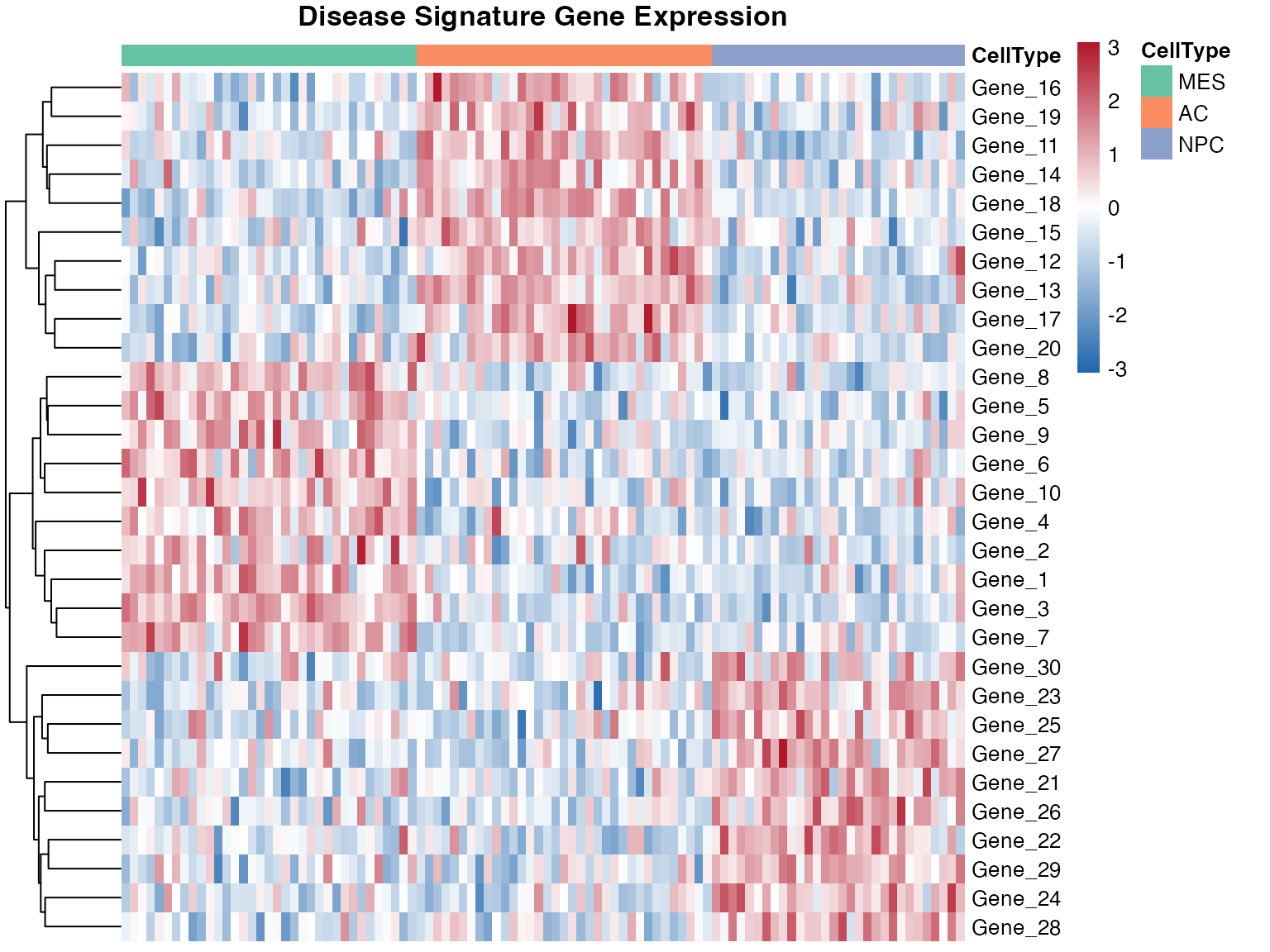
Disease Signature Heatmap
set.seed(123)
# Generate mock expression data
n_genes <- 30
n_cells <- 100
gene_names <- paste0("Gene_", 1:n_genes)
cell_types <- rep(c("MES", "AC", "NPC"), c(35, 35, 30))
# Create expression matrix with cell type-specific patterns
expr_matrix <- matrix(rnorm(n_genes * n_cells), nrow = n_genes, ncol = n_cells)
rownames(expr_matrix) <- gene_names
colnames(expr_matrix) <- paste0("Cell_", 1:n_cells)
# Add cell type-specific patterns
for (i in 1:10) {
expr_matrix[i, cell_types == "MES"] <- expr_matrix[i, cell_types == "MES"] + 1.5
}
for (i in 11:20) {
expr_matrix[i, cell_types == "AC"] <- expr_matrix[i, cell_types == "AC"] + 1.5
}
for (i in 21:30) {
expr_matrix[i, cell_types == "NPC"] <- expr_matrix[i, cell_types == "NPC"] + 1.5
}
# Annotation
annotation_col <- data.frame(
CellType = factor(cell_types),
row.names = colnames(expr_matrix)
)
annotation_colors <- list(
CellType = c(MES = "#66C2A5", AC = "#FC8D62", NPC = "#8DA0CB")
)
pheatmap(
expr_matrix,
scale = "row",
show_colnames = FALSE,
annotation_col = annotation_col,
annotation_colors = annotation_colors,
color = colorRampPalette(c("#2166AC", "white", "#B2182B"))(100),
main = "Disease Signature Gene Expression",
fontsize = 10,
cluster_cols = FALSE
)
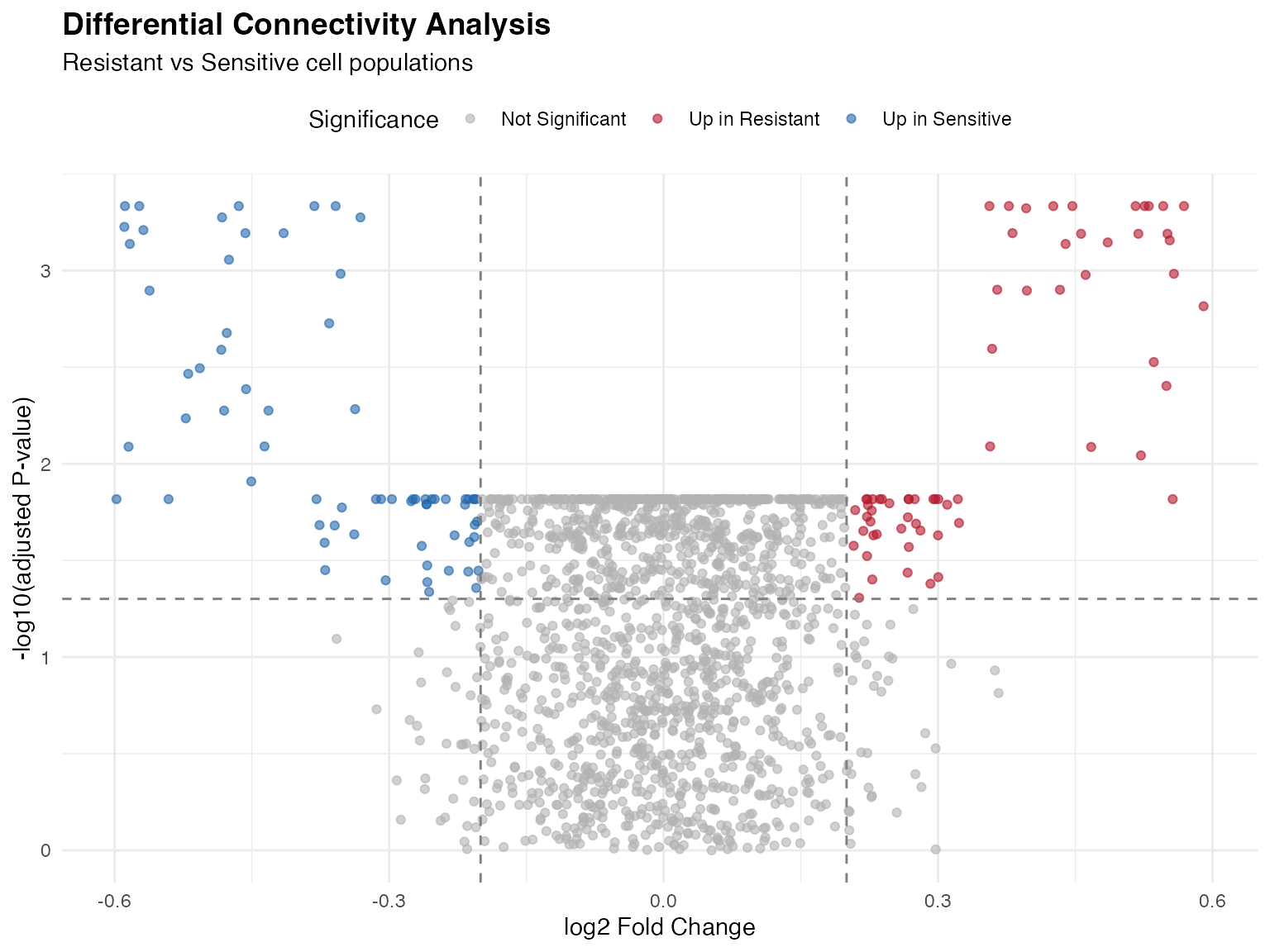
4. Volcano Plots
Differential Connectivity Volcano
set.seed(789)
n_compounds <- 1679
compound_names <- c("Temozolomide", "Vincristine", "Doxorubicin", "Erlotinib",
"Dasatinib", "Imatinib", "Sorafenib", "Sunitinib")
volcano_data <- data.frame(
Compound = paste0("Compound_", 1:n_compounds),
logFC = rnorm(n_compounds, 0, 0.12),
P.Value = 10^(-runif(n_compounds, 0, 3))
)
# Add significant hits
sig_up <- sample(1:n_compounds, 30)
sig_down <- sample(setdiff(1:n_compounds, sig_up), 30)
volcano_data$logFC[sig_up] <- runif(30, 0.3, 0.6)
volcano_data$logFC[sig_down] <- runif(30, -0.6, -0.3)
volcano_data$P.Value[c(sig_up, sig_down)] <- 10^(-runif(60, 3, 6))
volcano_data$adj.P.Val <- p.adjust(volcano_data$P.Value, method = "fdr")
volcano_data$Significance <- case_when(
volcano_data$logFC > 0.2 & volcano_data$adj.P.Val < 0.05 ~ "Up in Resistant",
volcano_data$logFC < -0.2 & volcano_data$adj.P.Val < 0.05 ~ "Up in Sensitive",
TRUE ~ "Not Significant"
)
# Add compound labels for top hits
volcano_data$Label <- ""
top_sig <- which(volcano_data$adj.P.Val < 0.001)
volcano_data$Label[top_sig[1:min(8, length(top_sig))]] <- compound_names[1:min(8, length(top_sig))]
ggplot(volcano_data, aes(x = logFC, y = -log10(adj.P.Val))) +
geom_point(aes(color = Significance), alpha = 0.6, size = 1.5) +
geom_hline(yintercept = -log10(0.05), linetype = "dashed", color = "gray50") +
geom_vline(xintercept = c(-0.2, 0.2), linetype = "dashed", color = "gray50") +
scale_color_manual(values = c(
"Up in Resistant" = "#B2182B",
"Up in Sensitive" = "#2166AC",
"Not Significant" = "gray70"
)) +
labs(
title = "Differential Connectivity Analysis",
subtitle = "Resistant vs Sensitive cell populations",
x = "log2 Fold Change",
y = "-log10(adjusted P-value)"
) +
theme_minimal() +
theme(
legend.position = "top",
plot.title = element_text(face = "bold", size = 14)
)
5. Reversal Score Visualization
Bar Plot of Top Reversal Compounds
reversal_data <- data.frame(
Compound = compound_names[1:8],
Reversal_Score = c(2.8, 2.5, 2.3, 2.1, 1.9, 1.8, 1.7, 1.6),
MOA = c("Kinase Inhibitor", "DNA Damage", "Epigenetic", "Metabolic",
"Kinase Inhibitor", "DNA Damage", "Epigenetic", "Other")
)
ggplot(reversal_data, aes(x = reorder(Compound, Reversal_Score), y = Reversal_Score, fill = MOA)) +
geom_col(width = 0.7) +
geom_hline(yintercept = 1, linetype = "dashed", color = "gray40") +
coord_flip() +
scale_fill_brewer(palette = "Set2", name = "Mechanism\nof Action") +
labs(
title = "Disease Signature Reversal Scores",
subtitle = "Top compounds for Mesenchymal state reversal",
x = "",
y = "Reversal Score (Discordant / Concordant)"
) +
theme_minimal() +
theme(
plot.title = element_text(face = "bold", size = 14),
legend.position = "right"
) +
annotate("text", x = 2, y = 1.5, label = "Reversal threshold",
color = "gray40", fontface = "italic", size = 3)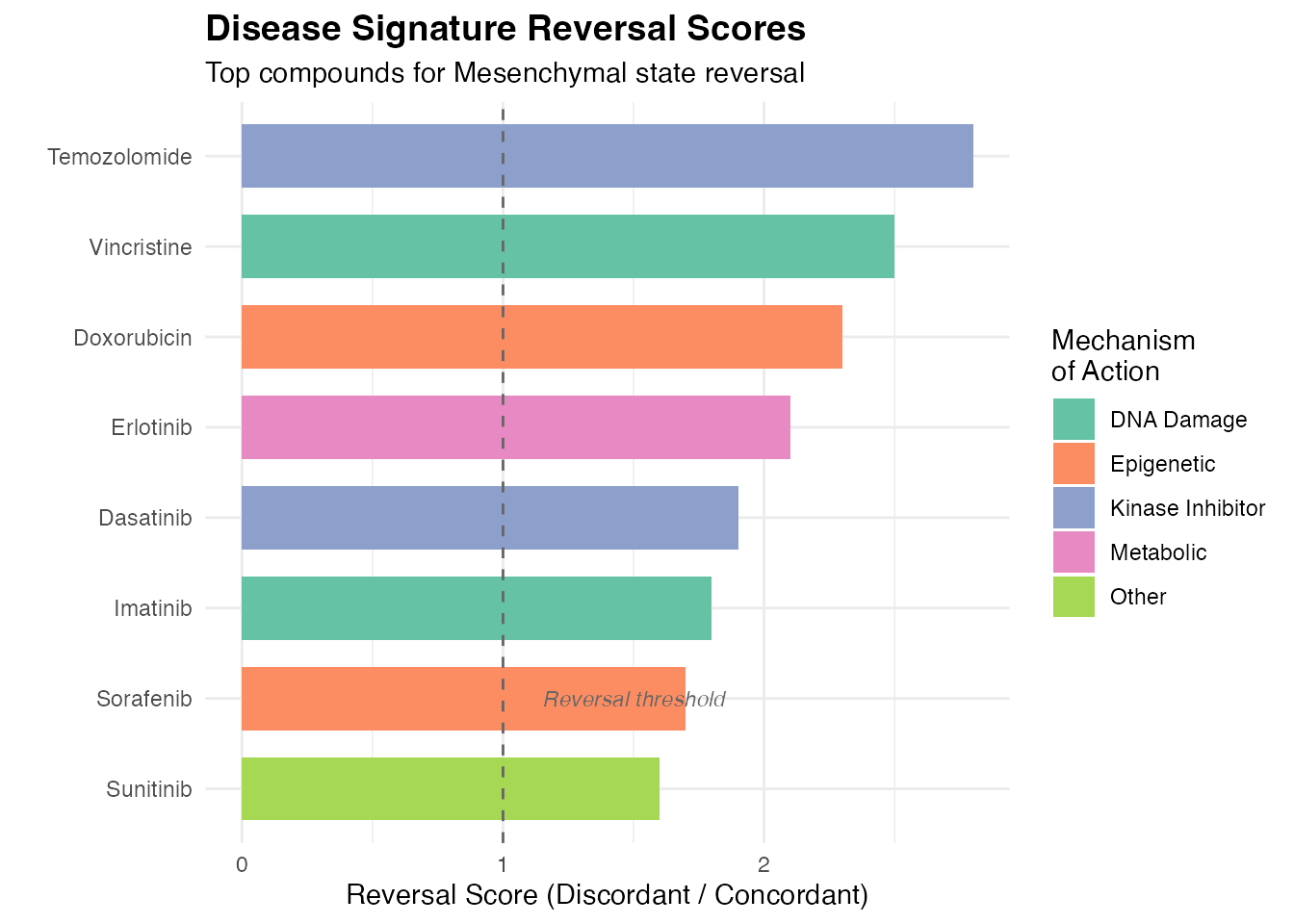
6. Multi-Panel Publication Figure
# Panel A: UMAP
p1 <- ggplot(umap_data, aes(x = UMAP1, y = UMAP2, color = Connectivity)) +
geom_point(size = 0.5, alpha = 0.6) +
scale_color_gradient2(low = "#2166AC", mid = "white", high = "#B2182B", midpoint = 0) +
labs(title = "A) Drug Connectivity", x = "UMAP 1", y = "UMAP 2") +
theme_minimal() +
theme(legend.position = "bottom", plot.title = element_text(face = "bold"))
# Panel B: Violin
p2 <- ggplot(tumor_data, aes(x = CellType, y = Connectivity, fill = CellType)) +
geom_violin(alpha = 0.7) +
geom_boxplot(width = 0.1, fill = "white") +
geom_hline(yintercept = 0, linetype = "dashed") +
scale_fill_brewer(palette = "Set2") +
labs(title = "B) By Cell Type", x = "", y = "Connectivity") +
theme_minimal() +
theme(legend.position = "none",
axis.text.x = element_text(angle = 30, hjust = 1),
plot.title = element_text(face = "bold"))
# Panel C: Volcano (subset)
p3 <- ggplot(volcano_data, aes(x = logFC, y = -log10(adj.P.Val), color = Significance)) +
geom_point(alpha = 0.5, size = 1) +
geom_hline(yintercept = -log10(0.05), linetype = "dashed") +
geom_vline(xintercept = c(-0.2, 0.2), linetype = "dashed") +
scale_color_manual(values = c("Up in Resistant" = "#B2182B",
"Up in Sensitive" = "#2166AC",
"Not Significant" = "gray70")) +
labs(title = "C) Differential Connectivity", x = "logFC", y = "-log10(adj.P)") +
theme_minimal() +
theme(legend.position = "bottom", plot.title = element_text(face = "bold"))
# Panel D: Reversal
p4 <- ggplot(reversal_data, aes(x = reorder(Compound, Reversal_Score), y = Reversal_Score)) +
geom_col(fill = "steelblue", width = 0.7) +
geom_hline(yintercept = 1, linetype = "dashed") +
coord_flip() +
labs(title = "D) Top Reversal Compounds", x = "", y = "Reversal Score") +
theme_minimal() +
theme(plot.title = element_text(face = "bold"))
gridExtra::grid.arrange(p1, p2, p3, p4, ncol = 2, nrow = 2)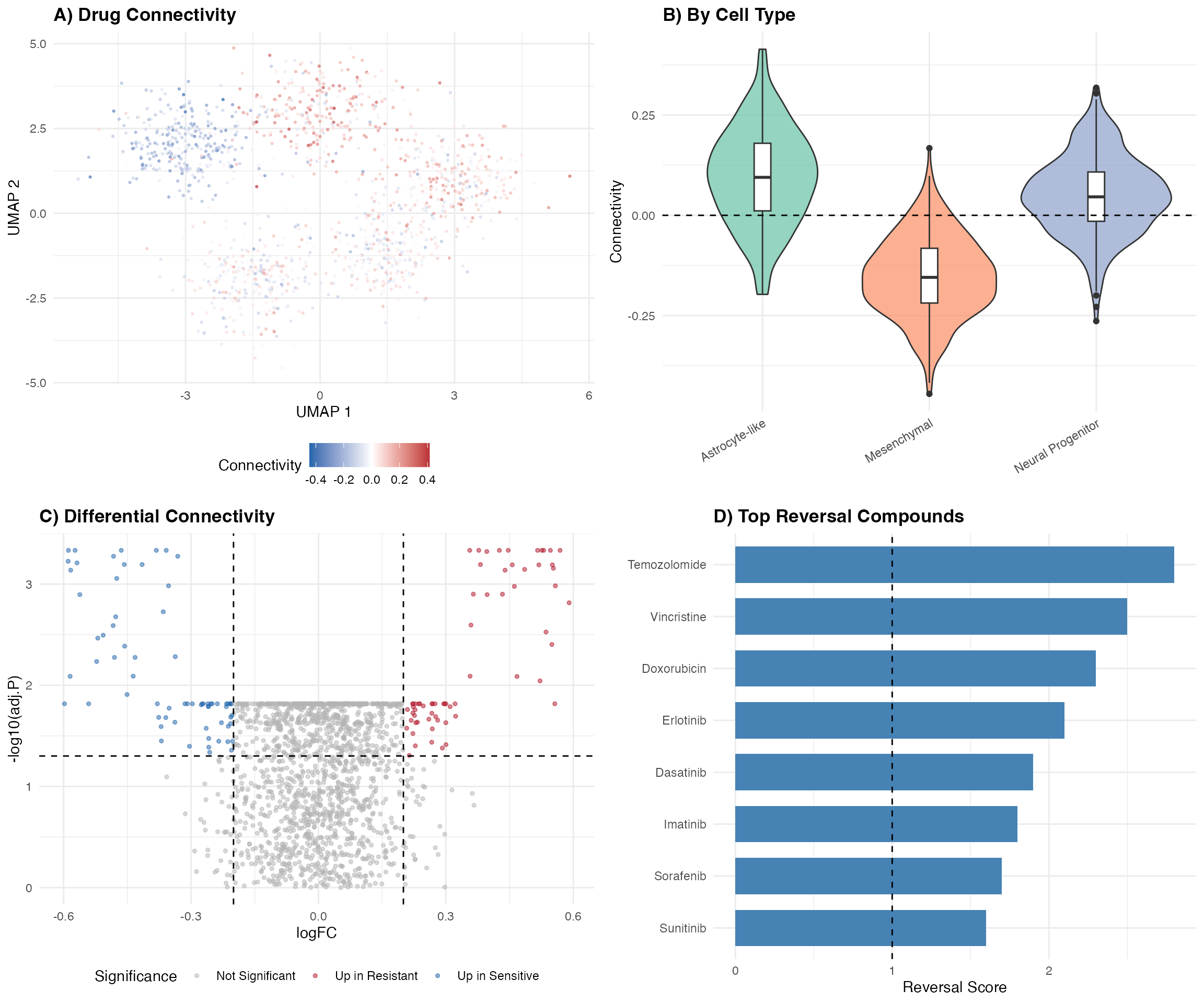
Customization Tips
Session Info
## R version 4.4.0 (2024-04-24)
## Platform: aarch64-apple-darwin20
## Running under: macOS 15.6.1
##
## Matrix products: default
## BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
##
## locale:
## [1] C
##
## time zone: Asia/Shanghai
## tzcode source: internal
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] pheatmap_1.0.13 viridis_0.6.5 viridisLite_0.4.2 tidyr_1.3.2
## [5] dplyr_1.1.4 ggplot2_4.0.1
##
## loaded via a namespace (and not attached):
## [1] gtable_0.3.6 jsonlite_2.0.0 compiler_4.4.0 tidyselect_1.2.1
## [5] dichromat_2.0-0.1 gridExtra_2.3 jquerylib_0.1.4 systemfonts_1.3.1
## [9] scales_1.4.0 textshaping_1.0.4 yaml_2.3.12 fastmap_1.2.0
## [13] R6_2.6.1 labeling_0.4.3 generics_0.1.4 knitr_1.51
## [17] htmlwidgets_1.6.4 tibble_3.3.1 desc_1.4.3 bslib_0.9.0
## [21] pillar_1.11.1 RColorBrewer_1.1-3 rlang_1.1.7 cachem_1.1.0
## [25] xfun_0.56 fs_1.6.6 sass_0.4.10 S7_0.2.1
## [29] otel_0.2.0 cli_3.6.5 pkgdown_2.2.0 withr_3.0.2
## [33] magrittr_2.0.4 digest_0.6.39 grid_4.4.0 lifecycle_1.0.5
## [37] vctrs_0.7.1 evaluate_1.0.5 glue_1.8.0 farver_2.1.2
## [41] ragg_1.5.0 purrr_1.2.1 rmarkdown_2.30 tools_4.4.0
## [45] pkgconfig_2.0.3 htmltools_0.5.9