Quick Start Guide
Zaoqu Liu, Robert K. Suter, Nagi G. Ayad
2026-02-03
Source:vignettes/quick-start.Rmd
quick-start.RmdIntroduction
scFOCAL (Single-Cell Framework for Omics-Connectivity and Analysis via L1000) is a computational framework designed to bridge single-cell transcriptomics with pharmacological knowledge. By integrating drug-response transcriptional consensus signatures (TCS) from the LINCS L1000 database with single-cell RNA sequencing data, scFOCAL enables:
- Identification of drug-sensitive and drug-resistant cell populations
- Discovery of combination therapy candidates
- Analysis of tumor heterogeneity in drug response
This vignette provides a quick introduction to get you started with scFOCAL.
Installation
From R-Universe (Recommended)
install.packages("scFOCAL", repos = "https://zaoqu-liu.r-universe.dev")From GitHub
# Install devtools if needed
if (!requireNamespace("devtools", quietly = TRUE))
install.packages("devtools")
devtools::install_github("Zaoqu-Liu/scFOCAL")Dependencies
scFOCAL requires several Bioconductor packages:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c("MAST", "edgeR", "ComplexHeatmap", "EnhancedVolcano"))Note: Seurat is required but listed as a suggested package:
# For Seurat v4 (recommended)
remotes::install_version("Seurat", version = "4.4.0")
# Or for Seurat v5
install.packages("Seurat")Launching scFOCAL
Once installed, launching the interactive Shiny application is straightforward:
library(scFOCAL)
# Launch the GUI
runscFOCAL()This will open the scFOCAL interface in your default web browser.
Workflow Overview
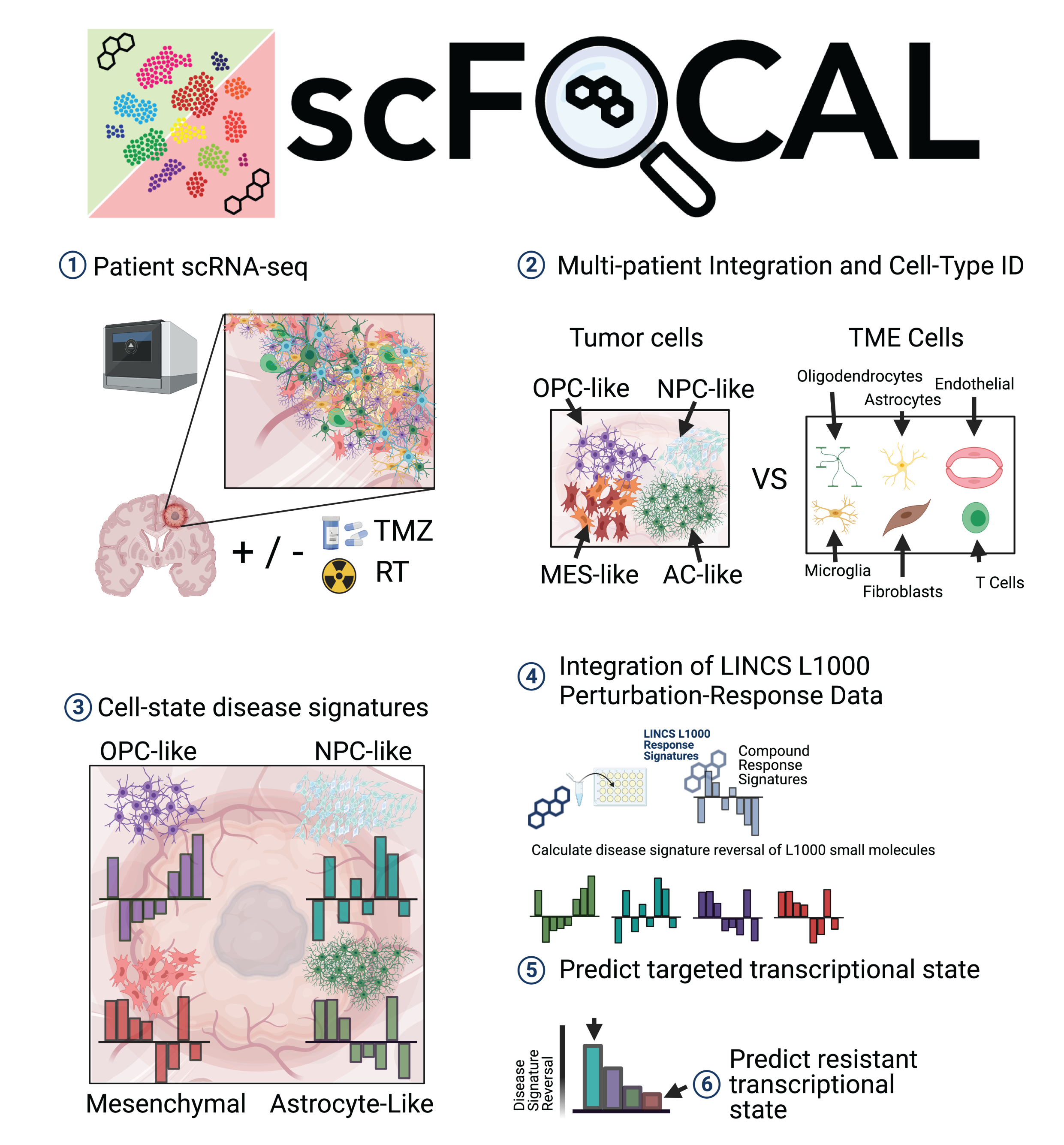
The scFOCAL workflow consists of five main steps:
Step 1: Data Upload
Upload your preprocessed Seurat object (.rds format)
containing:
- Normalized expression data
- Cell type annotations in metadata
- Dimensional reduction (UMAP/tSNE)

Step 2: Pre-processing
Define your analysis groups:
- Control populations: Non-malignant cells (e.g., immune cells, stromal cells)
- Test populations: Tumor cells of interest

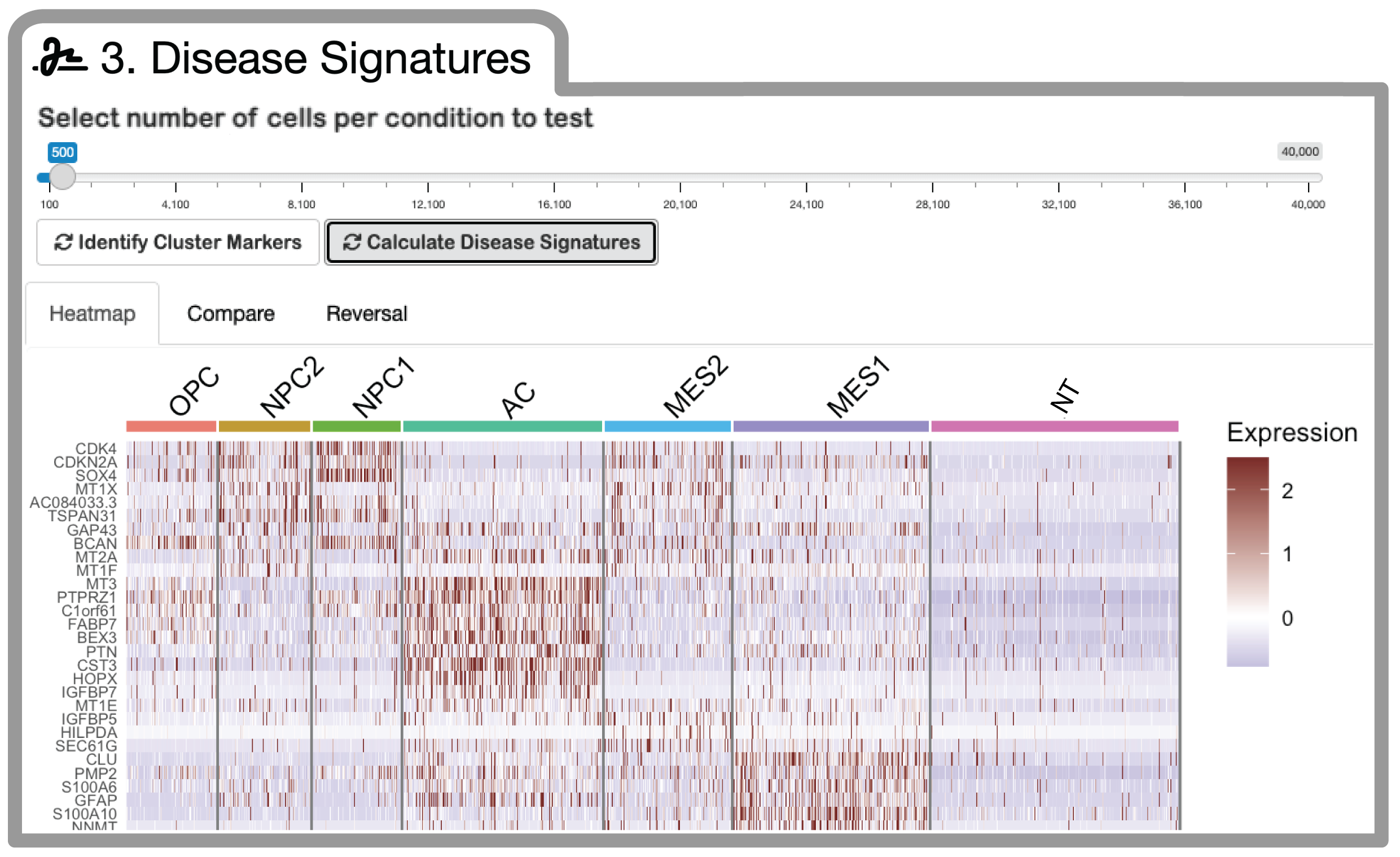
Step 3: Disease Signature Generation
Compute cell-type-specific differential expression signatures using the MAST statistical framework:
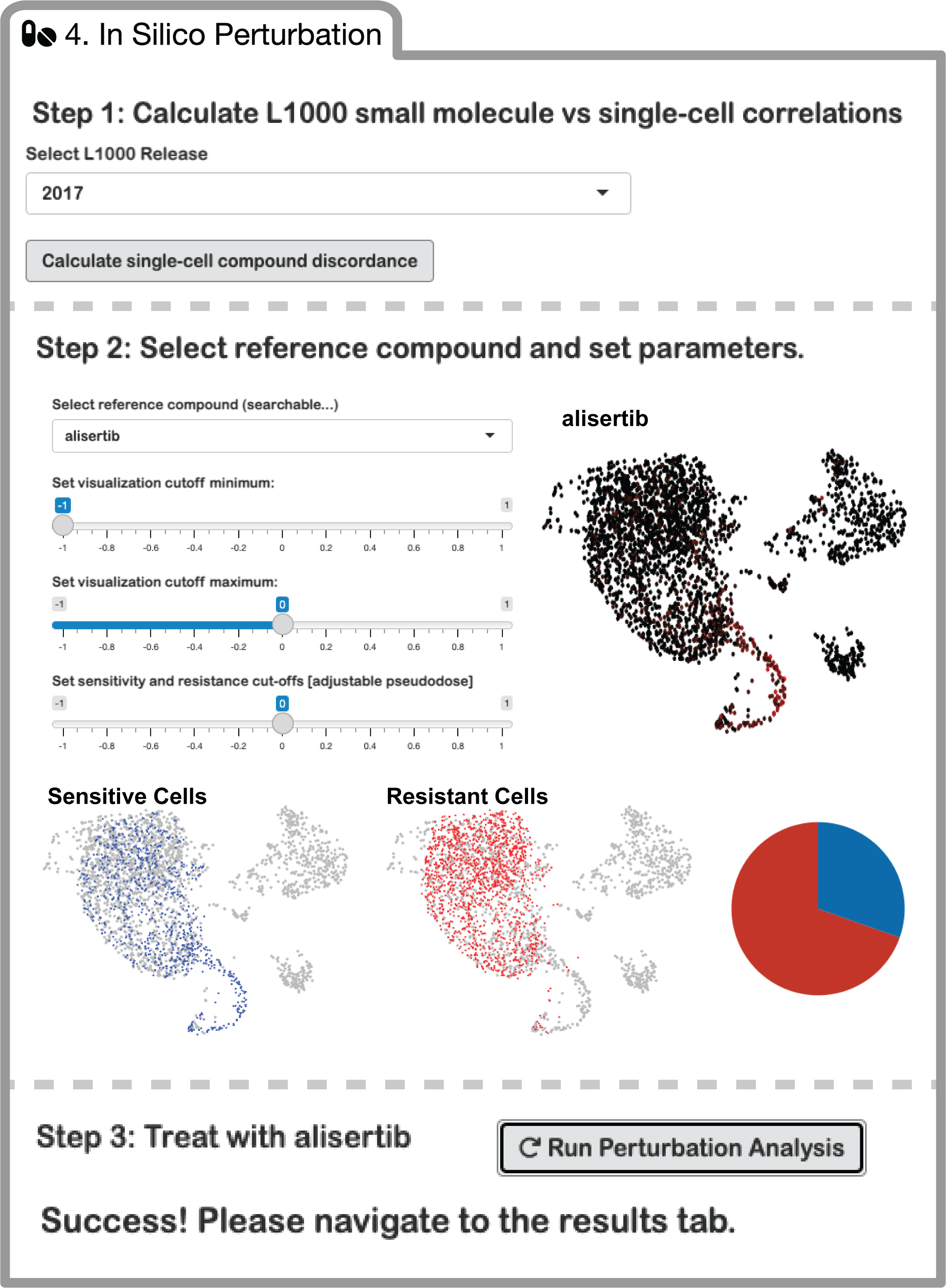
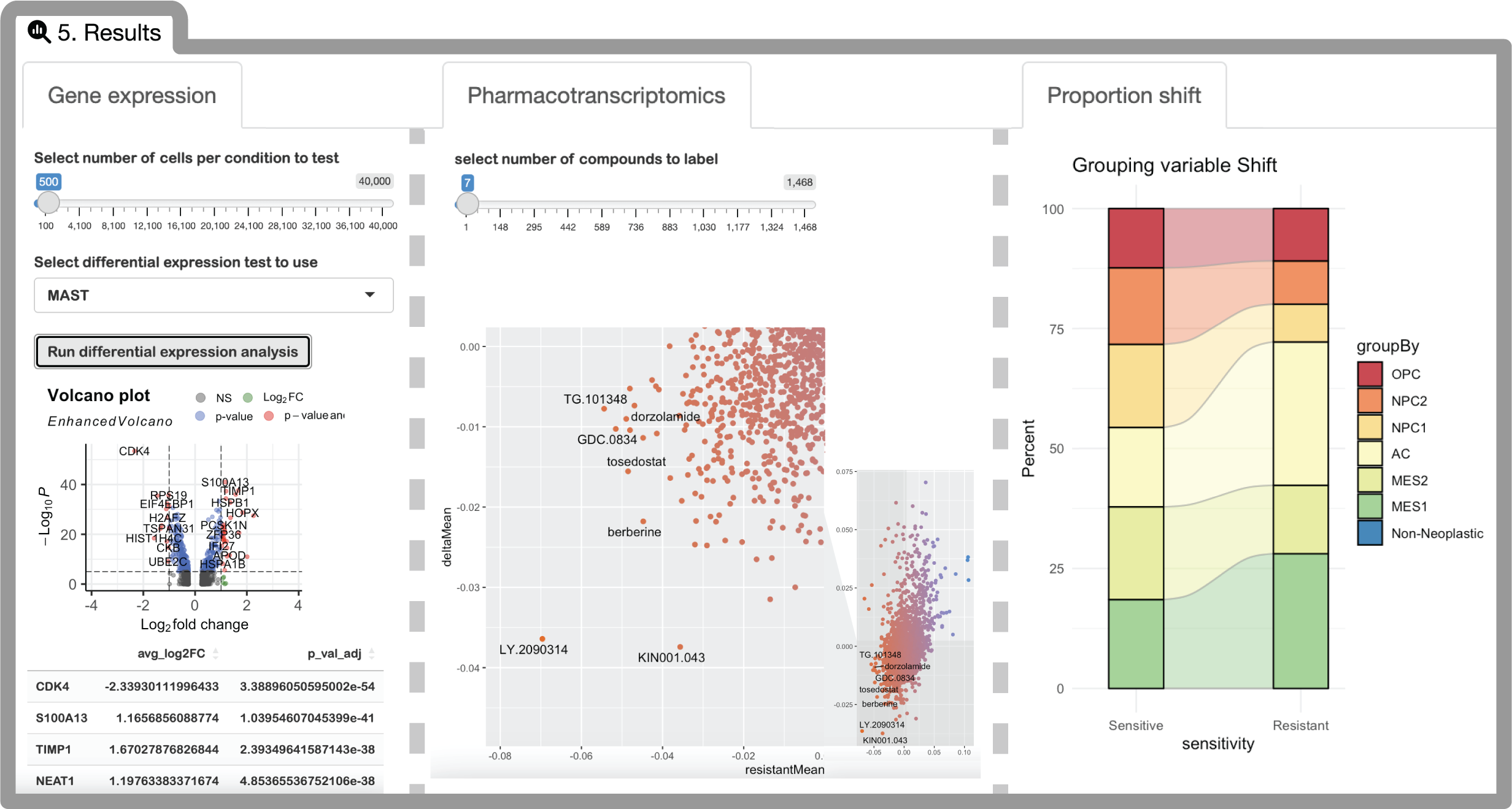
Next Steps
For more detailed information, see:
- Algorithm Principles - Mathematical foundations
- Statistical Framework - Statistical methods
- Case Study: GBM Analysis - Complete analysis example
- Visualization Gallery - Output visualizations
Citation
If you use scFOCAL in your research, please cite:
Suter RK, Jermakowicz AM, Veeramachaneni R, et al. Drug and single-cell gene expression integration identifies sensitive and resistant glioblastoma cell populations. Nature Communications 17, 99 (2026). https://doi.org/10.1038/s41467-025-67783-5
Session Info
## R version 4.4.0 (2024-04-24)
## Platform: aarch64-apple-darwin20
## Running under: macOS 15.6.1
##
## Matrix products: default
## BLAS: /System/Library/Frameworks/Accelerate.framework/Versions/A/Frameworks/vecLib.framework/Versions/A/libBLAS.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
##
## locale:
## [1] C
##
## time zone: Asia/Shanghai
## tzcode source: internal
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## loaded via a namespace (and not attached):
## [1] digest_0.6.39 desc_1.4.3 R6_2.6.1 fastmap_1.2.0
## [5] xfun_0.56 cachem_1.1.0 knitr_1.51 htmltools_0.5.9
## [9] rmarkdown_2.30 lifecycle_1.0.5 cli_3.6.5 sass_0.4.10
## [13] pkgdown_2.2.0 textshaping_1.0.4 jquerylib_0.1.4 systemfonts_1.3.1
## [17] compiler_4.4.0 tools_4.4.0 ragg_1.5.0 bslib_0.9.0
## [21] evaluate_1.0.5 yaml_2.3.12 otel_0.2.0 jsonlite_2.0.0
## [25] rlang_1.1.7 fs_1.6.6 htmlwidgets_1.6.4