Dynamical Model Deep Dive
Zaoqu Liu
2026-01-26
Source:vignettes/dynamical-model.Rmd
dynamical-model.RmdIntroduction
The dynamical model in scVeloR represents the most sophisticated approach to RNA velocity estimation. Unlike simpler steady-state methods, it infers the full kinetic parameters of gene expression without assuming transcriptional equilibrium.
This vignette provides a comprehensive guide to:
- Understanding the underlying mathematics
- Implementing the dynamical model
- Interpreting results
- Troubleshooting common issues
Mathematical Foundation
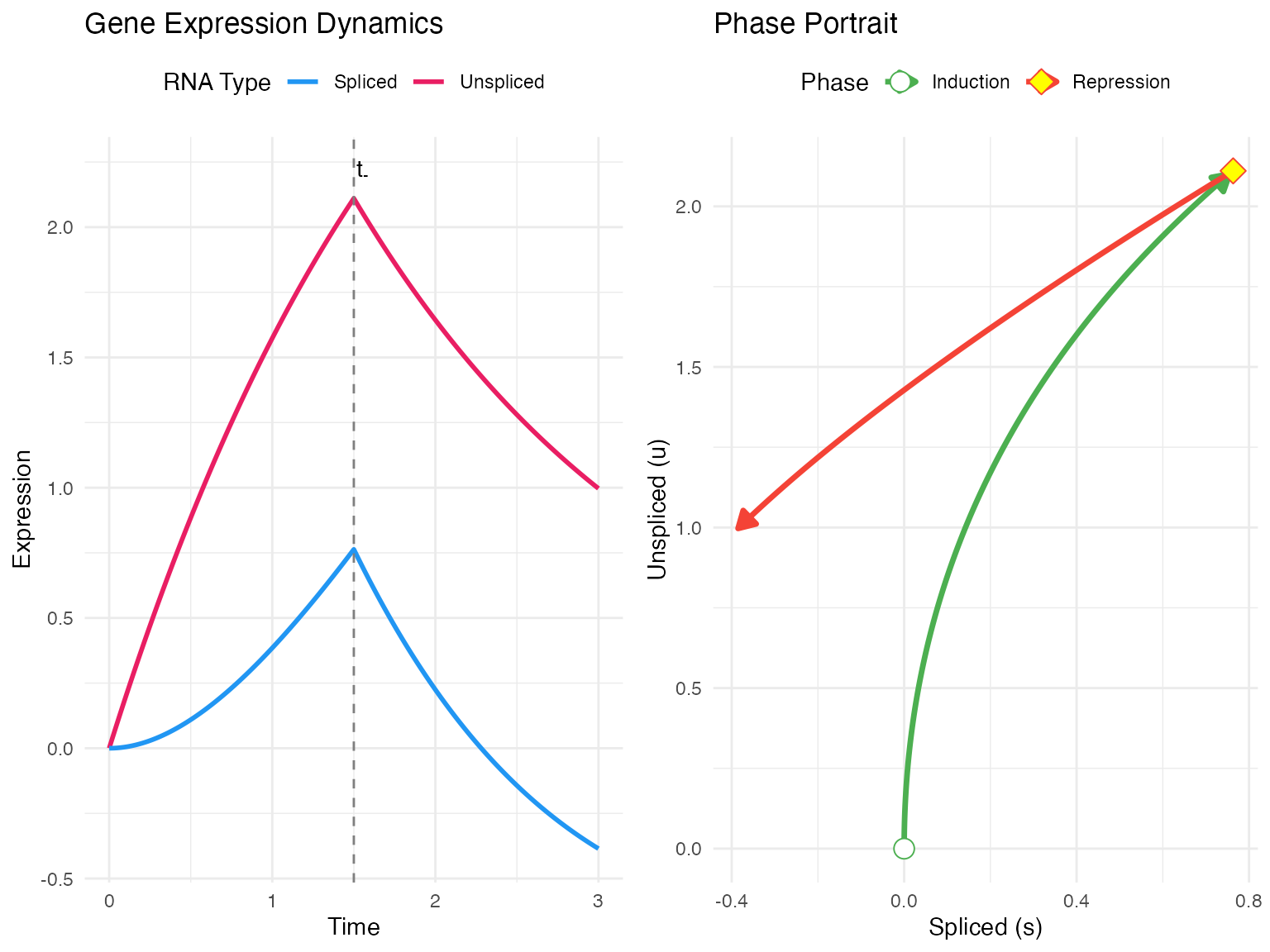
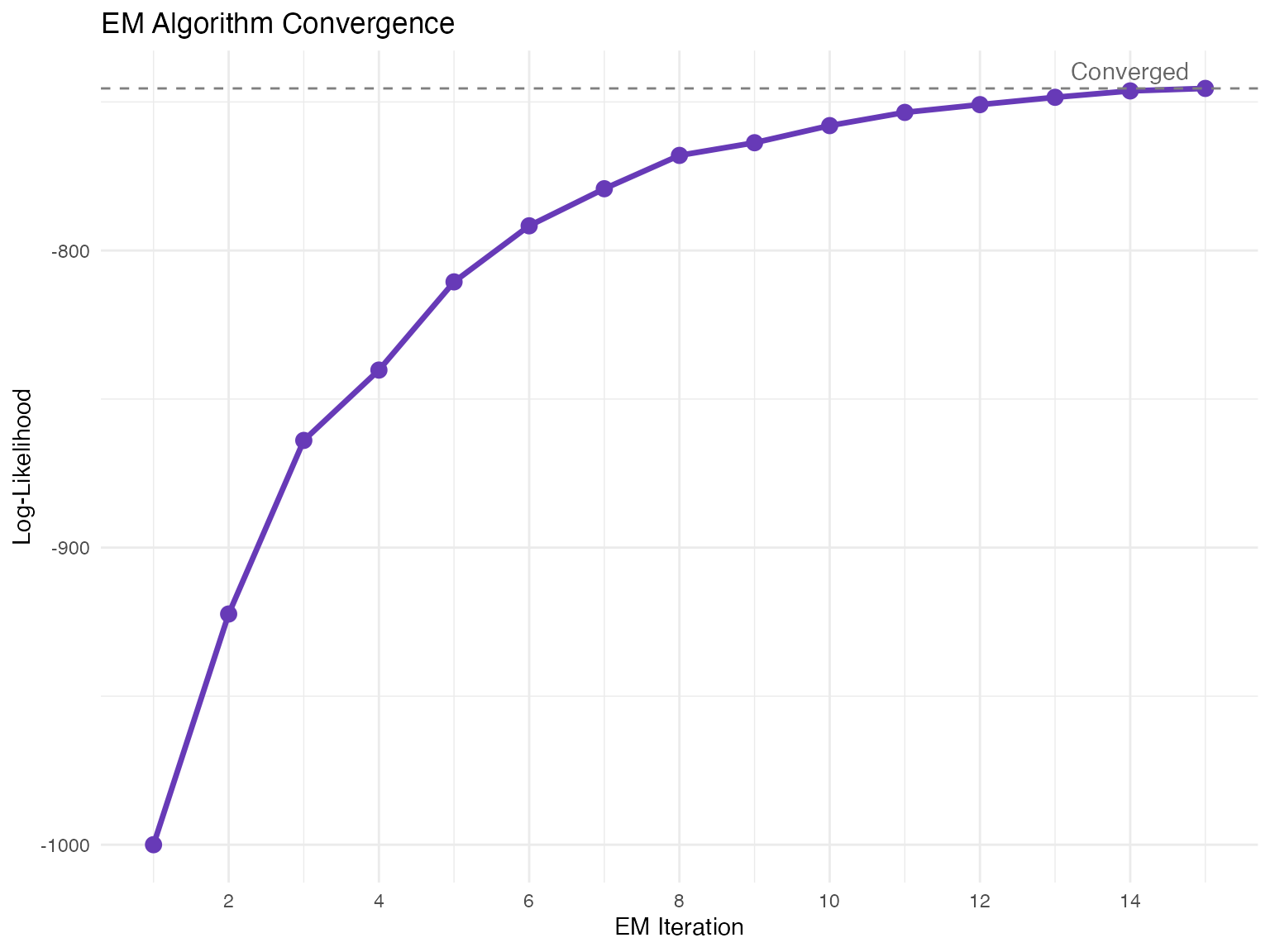
The EM Algorithm
Overview
The dynamical model uses an Expectation-Maximization (EM) algorithm to jointly infer:
- Kinetic parameters: , , for each gene
- Switching time: for each gene
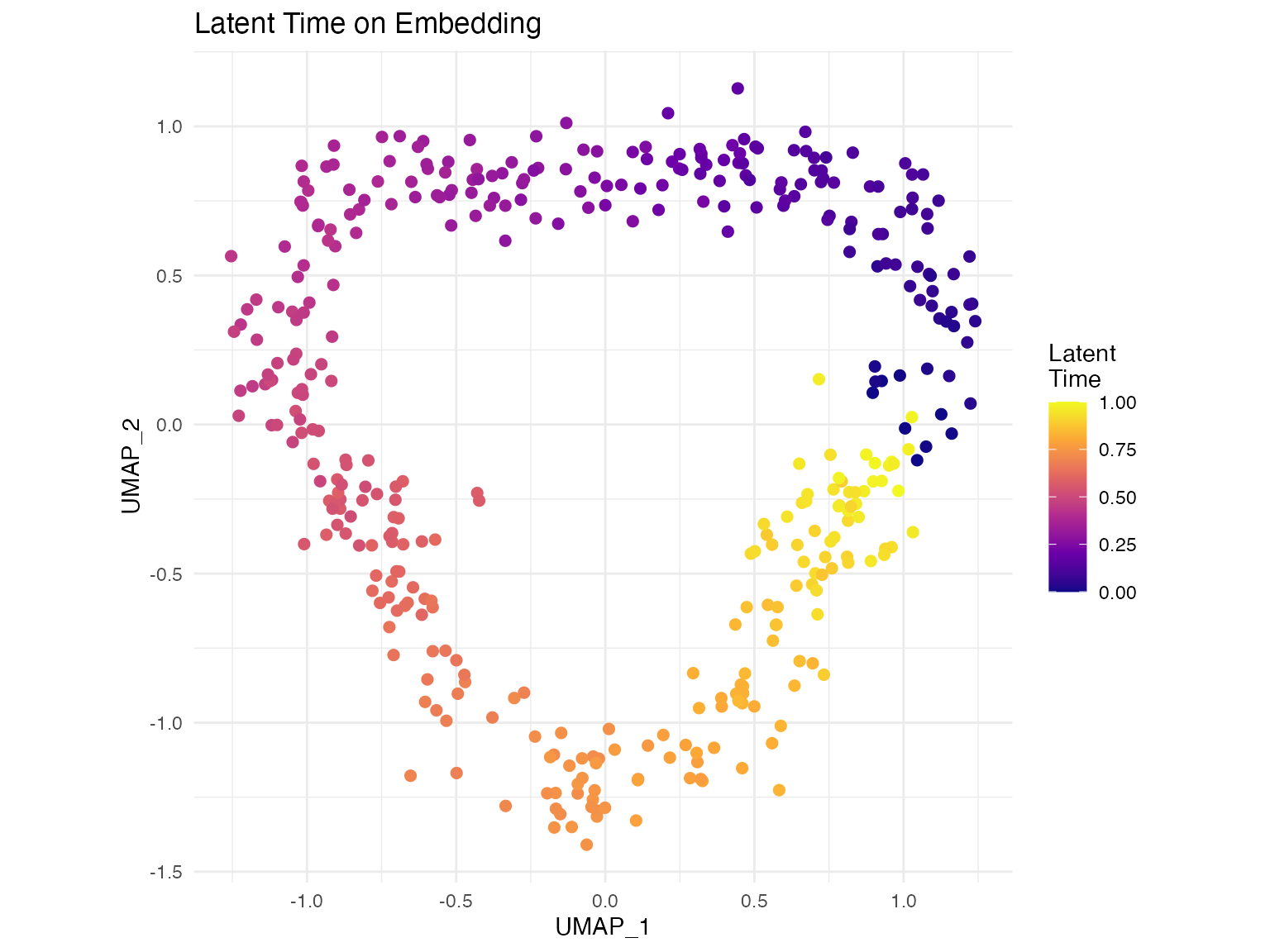
- Latent time: for each cell
Implementation in scVeloR
Basic Usage
library(scVeloR)
# Run dynamical model
seurat_obj <- velocity(seurat_obj,
mode = "dynamical",
max_iter = 10)Step-by-Step Approach
# Step 1: Identify velocity genes using steady-state
seurat_obj <- velocity(seurat_obj, mode = "deterministic")
# Step 2: Recover full dynamics
seurat_obj <- recover_dynamics(
seurat_obj,
genes = "velocity_genes", # Use pre-selected genes
n_top_genes = 2000, # Or top N by variance
max_iter = 10, # EM iterations
fit_scaling = TRUE, # Estimate scaling factor
n_cores = 4 # Parallel processing
)
# Step 3: Compute velocity from dynamics
seurat_obj <- velocity_from_dynamics(seurat_obj)
# Step 4: Compute latent time
seurat_obj <- compute_latent_time(seurat_obj)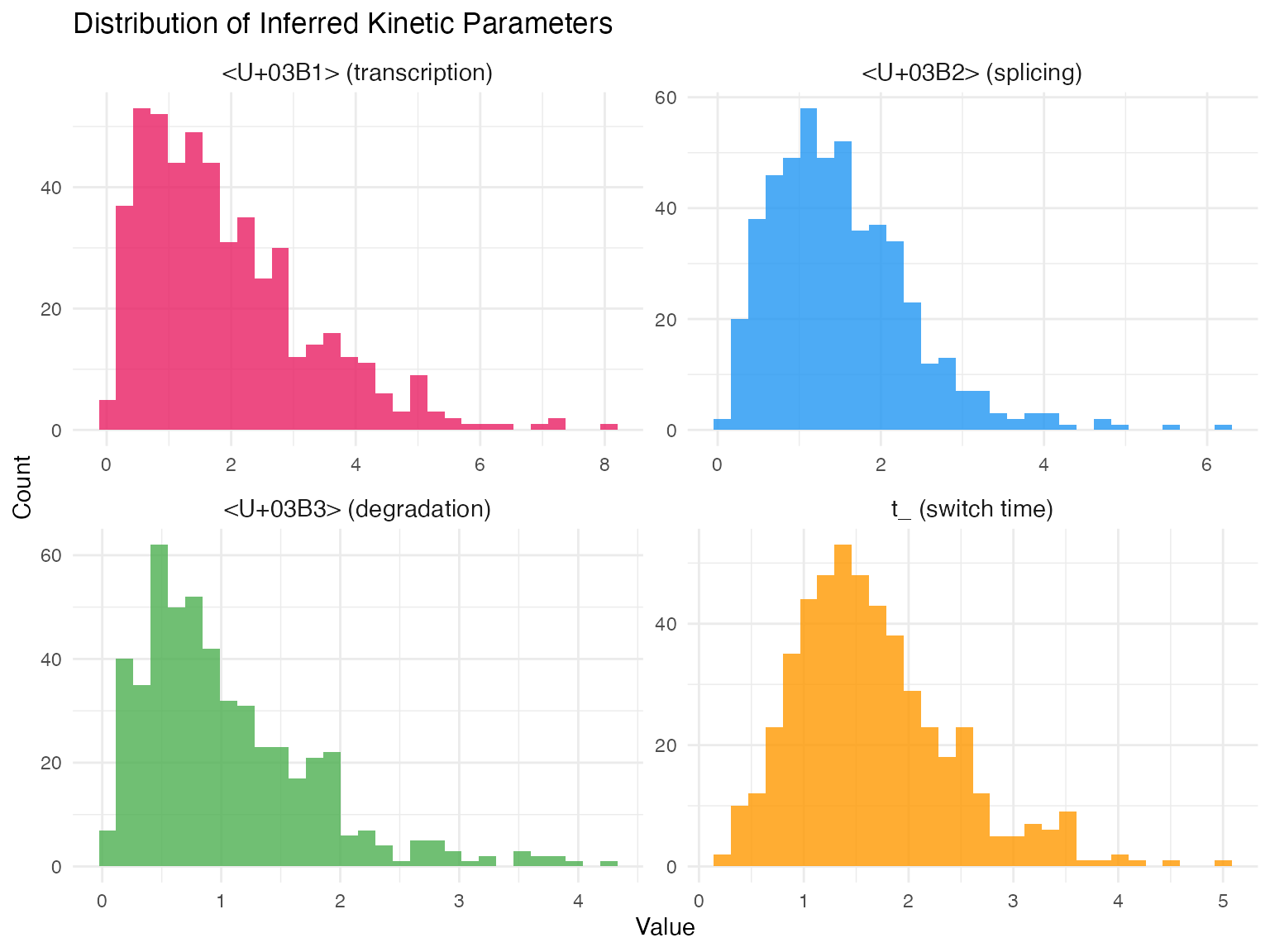
Quality Control
Velocity Confidence
# Compute velocity confidence
seurat_obj <- velocity_confidence(seurat_obj)
# Check distribution
hist(seurat_obj$velocity_confidence, breaks = 50,
main = "Velocity Confidence Distribution",
xlab = "Confidence Score")Troubleshooting
Common Issues
-
Poor convergence
- Increase
max_iter - Filter low-quality genes
- Check data normalization
- Increase
-
Unrealistic parameter estimates
- Check for outlier cells
- Verify spliced/unspliced layer quality
- Try different gene selection
-
Inconsistent latent time
- Use more genes for averaging
- Filter genes with low likelihood
- Check for batch effects
Best Practices
# 1. Pre-filter low-quality data
seurat_obj <- prepare_velocity(seurat_obj,
min_counts = 20,
min_cells = 30)
# 2. Use sufficient EM iterations
seurat_obj <- velocity(seurat_obj,
mode = "dynamical",
max_iter = 15) # More iterations
# 3. Check results
velocity_summary(seurat_obj)
# 4. Filter by gene quality
gene_params <- seurat_obj@misc$scVeloR$dynamics$gene_params
good_fit <- gene_params$likelihood > 0.2
message(sprintf("%d / %d genes with good fit", sum(good_fit), length(good_fit)))Advanced Topics
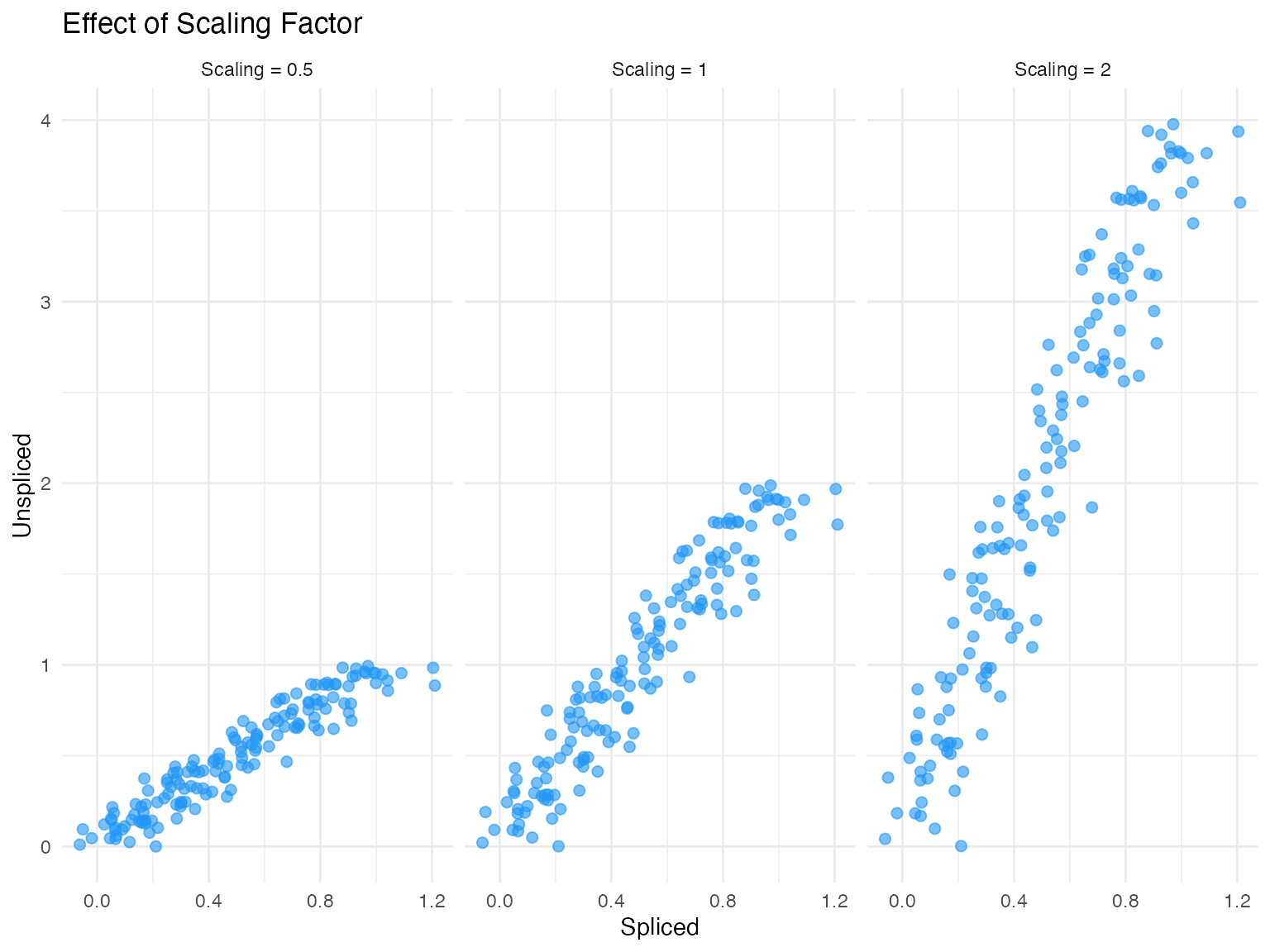
Summary
The dynamical model provides the most comprehensive RNA velocity analysis by:
- Inferring full kinetics: α, β, γ for each gene
- Handling transient states: No equilibrium assumption
- Providing latent time: Absolute temporal ordering of cells
Use it when you need: - Publication-quality results - Analysis of rapidly changing processes - Interpretation of kinetic parameters - Latent time for downstream analysis
Session Information
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] tidyr_1.3.2 gridExtra_2.3 ggplot2_4.0.1
#>
#> loaded via a namespace (and not attached):
#> [1] gtable_0.3.6 jsonlite_2.0.0 dplyr_1.1.4 compiler_4.4.0
#> [5] tidyselect_1.2.1 dichromat_2.0-0.1 jquerylib_0.1.4 systemfonts_1.3.1
#> [9] scales_1.4.0 textshaping_1.0.4 yaml_2.3.12 fastmap_1.2.0
#> [13] R6_2.6.1 labeling_0.4.3 generics_0.1.4 knitr_1.51
#> [17] htmlwidgets_1.6.4 tibble_3.3.1 desc_1.4.3 bslib_0.9.0
#> [21] pillar_1.11.1 RColorBrewer_1.1-3 rlang_1.1.7 cachem_1.1.0
#> [25] xfun_0.56 fs_1.6.6 sass_0.4.10 S7_0.2.1
#> [29] otel_0.2.0 viridisLite_0.4.2 cli_3.6.5 pkgdown_2.1.3
#> [33] withr_3.0.2 magrittr_2.0.4 digest_0.6.39 grid_4.4.0
#> [37] lifecycle_1.0.5 vctrs_0.7.1 evaluate_1.0.5 glue_1.8.0
#> [41] farver_2.1.2 ragg_1.5.0 purrr_1.2.1 rmarkdown_2.30
#> [45] tools_4.4.0 pkgconfig_2.0.3 htmltools_0.5.9