Case Study: Hematopoiesis
Analyzing Blood Cell Development with CellOracleR
Zaoqu Liu
2026-01-25
Source:vignettes/case-study.Rmd
case-study.RmdIntroduction
This case study demonstrates a complete CellOracleR analysis workflow using hematopoiesis (blood cell development) as an example. We will simulate the effects of key transcription factor perturbations on cell fate decisions.
Biological Background
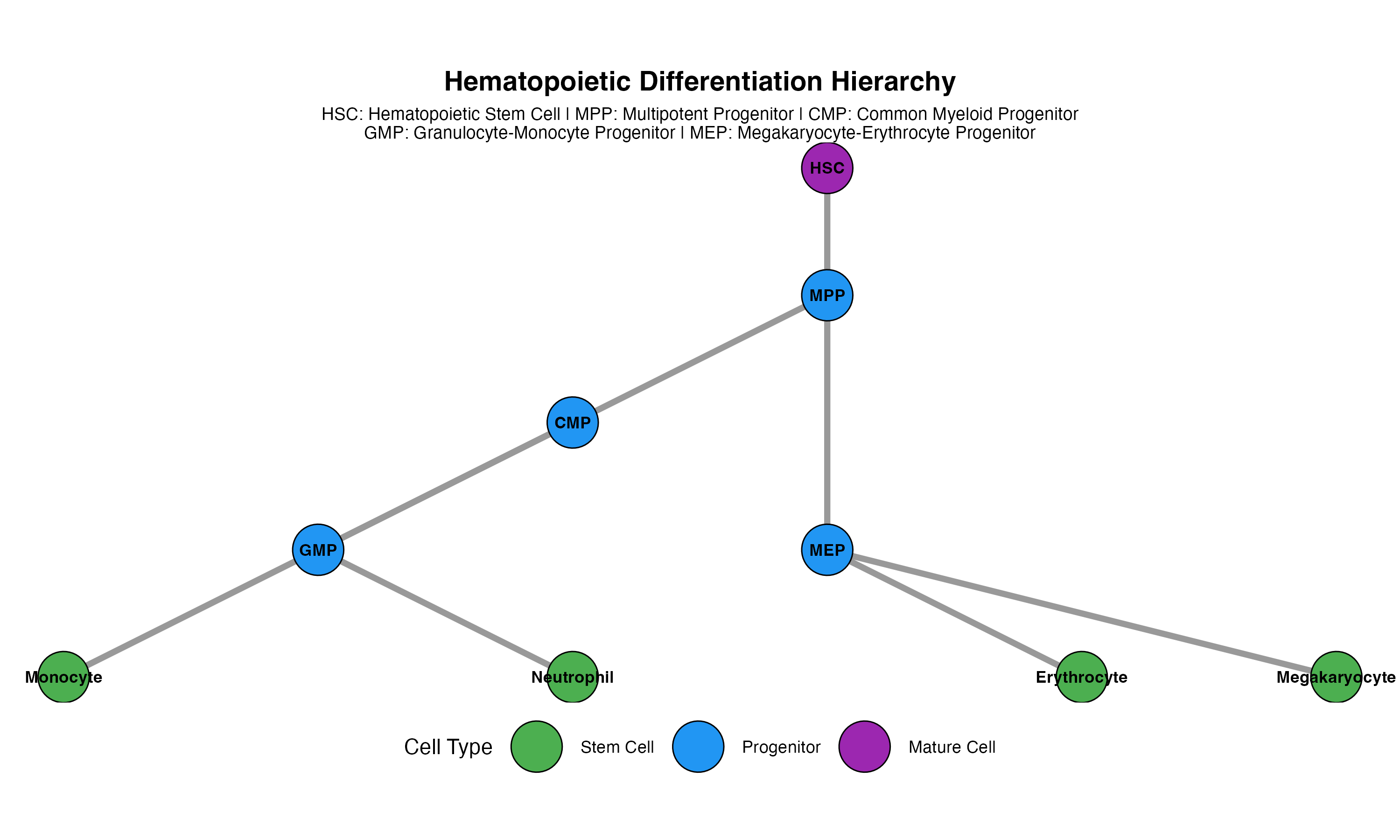
Simulated Data Setup
For demonstration, we create a simulated hematopoiesis dataset:
set.seed(42)
# Simulate cells along differentiation trajectories
n_cells <- 2000
# Define cluster centers in UMAP space
cluster_centers <- list(
HSC = c(0, 3),
MPP = c(0, 2),
CMP = c(-1, 1),
GMP = c(-2, 0),
MEP = c(1, 0),
Monocyte = c(-3, -1),
Neutrophil = c(-1.5, -1),
Erythrocyte = c(2, -1),
Megakaryocyte = c(3, 0)
)
# Simulate cells for each cluster
cells_per_cluster <- c(200, 300, 250, 200, 250, 200, 200, 250, 150)
names(cells_per_cluster) <- names(cluster_centers)
embedding_list <- list()
for (i in seq_along(cluster_centers)) {
cluster_name <- names(cluster_centers)[i]
center <- cluster_centers[[i]]
n <- cells_per_cluster[i]
embedding_list[[i]] <- data.frame(
UMAP_1 = rnorm(n, center[1], 0.4),
UMAP_2 = rnorm(n, center[2], 0.4),
cell_type = cluster_name
)
}
demo_data <- do.call(rbind, embedding_list)
demo_data$cell_id <- paste0("cell_", 1:nrow(demo_data))
# Plot the data
ggplot(demo_data, aes(x = UMAP_1, y = UMAP_2, color = cell_type)) +
geom_point(alpha = 0.6, size = 1) +
scale_color_manual(values = c(
"HSC" = "#9C27B0",
"MPP" = "#7B1FA2",
"CMP" = "#1976D2",
"GMP" = "#0288D1",
"MEP" = "#0097A7",
"Monocyte" = "#388E3C",
"Neutrophil" = "#689F38",
"Erythrocyte" = "#F44336",
"Megakaryocyte" = "#E91E63"
)) +
labs(
title = "Simulated Hematopoiesis Dataset",
subtitle = paste(nrow(demo_data), "cells across 9 cell types"),
x = "UMAP 1",
y = "UMAP 2",
color = "Cell Type"
) +
theme_minimal() +
theme(
panel.grid = element_blank(),
plot.title = element_text(face = "bold", size = 14)
) +
coord_fixed()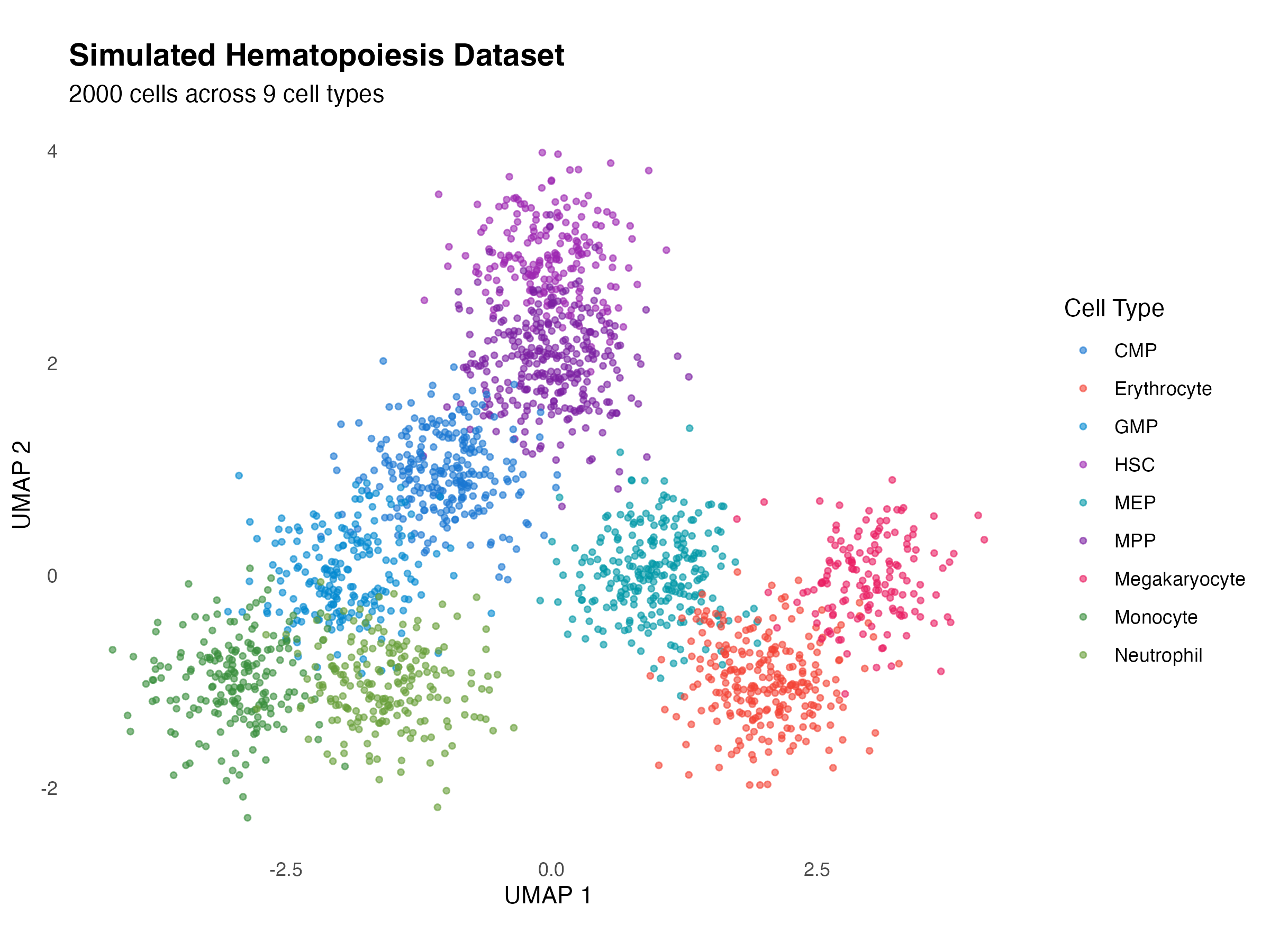
Analysis Workflow
Step 1: Create Oracle Object
library(CellOracleR)
library(Seurat)
# Assuming you have a Seurat object
oracle <- create_oracle(
seurat_obj = seurat_obj,
cluster_col = "cell_type",
embedding_name = "umap"
)
# View basic information
print(oracle)Step 2: Import Base GRN
# Load TF-target dictionary from motif analysis
base_grn <- load_base_grn("path/to/base_grn.csv")
# Or create from ATAC-seq + Cicero output
base_grn <- get_base_grn_from_cicero(
cicero_connections = cicero_output,
peak_gene_links = peak_gene_table
)
# Import to Oracle
oracle$import_TF_data(TFdict = base_grn)Step 3: Fit GRN
# Fit cluster-specific GRNs
oracle$fit_grn_for_simulation(
GRN_unit = "cluster",
alpha = 10,
bagging_number = 200
)Step 4: Simulate Perturbations
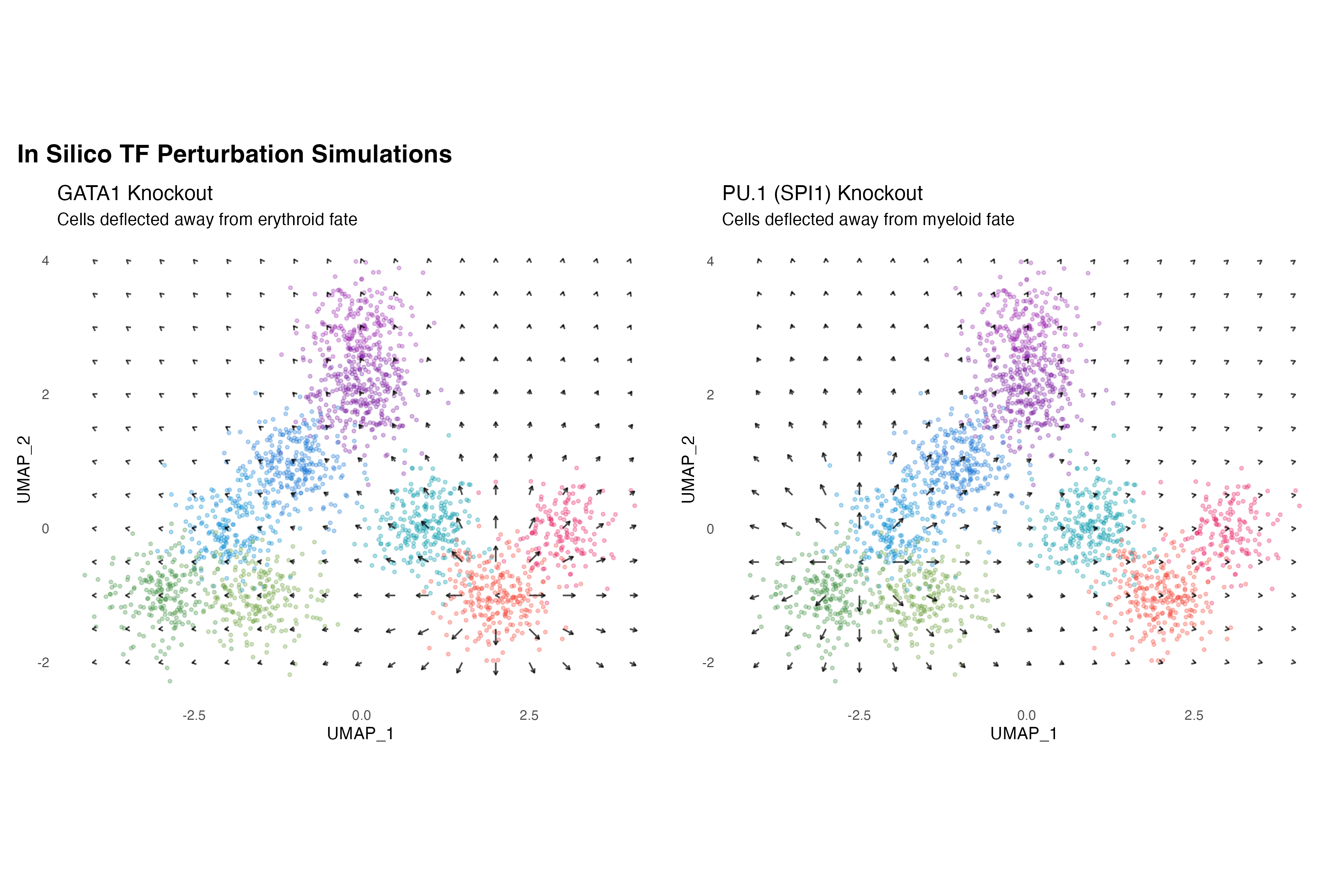
Let’s simulate the effects of knocking out key TFs:
# Simulate GATA1 knockout effect
# GATA1 drives erythroid differentiation, so KO should reduce erythroid cells
# Create simulated flow vectors
# For GATA1 KO: vectors should point away from erythroid fate
# Calculate grid
grid_x <- seq(-4, 4, by = 0.5)
grid_y <- seq(-2, 4, by = 0.5)
grid <- expand.grid(x = grid_x, y = grid_y)
# GATA1 KO: reduce flow toward erythroid
erythroid_center <- c(2, -1)
grid$dx_gata1ko <- 0
grid$dy_gata1ko <- 0
for (i in 1:nrow(grid)) {
# Calculate direction away from erythroid
dir_x <- grid$x[i] - erythroid_center[1]
dir_y <- grid$y[i] - erythroid_center[2]
dist <- sqrt(dir_x^2 + dir_y^2)
# Stronger effect near erythroid
strength <- exp(-dist/2) * 0.3
grid$dx_gata1ko[i] <- dir_x / max(dist, 0.1) * strength
grid$dy_gata1ko[i] <- dir_y / max(dist, 0.1) * strength
}
# PU.1 KO: reduce flow toward myeloid
myeloid_center <- c(-2.5, -0.5)
grid$dx_pu1ko <- 0
grid$dy_pu1ko <- 0
for (i in 1:nrow(grid)) {
dir_x <- grid$x[i] - myeloid_center[1]
dir_y <- grid$y[i] - myeloid_center[2]
dist <- sqrt(dir_x^2 + dir_y^2)
strength <- exp(-dist/2) * 0.3
grid$dx_pu1ko[i] <- dir_x / max(dist, 0.1) * strength
grid$dy_pu1ko[i] <- dir_y / max(dist, 0.1) * strength
}
# Create side-by-side plots
p1 <- ggplot() +
geom_point(data = demo_data,
aes(x = UMAP_1, y = UMAP_2, color = cell_type),
alpha = 0.3, size = 0.8) +
geom_segment(data = grid,
aes(x = x, y = y, xend = x + dx_gata1ko, yend = y + dy_gata1ko),
arrow = arrow(length = unit(0.1, "cm")),
color = "black", alpha = 0.7) +
scale_color_manual(values = c(
"HSC" = "#9C27B0", "MPP" = "#7B1FA2", "CMP" = "#1976D2",
"GMP" = "#0288D1", "MEP" = "#0097A7", "Monocyte" = "#388E3C",
"Neutrophil" = "#689F38", "Erythrocyte" = "#F44336", "Megakaryocyte" = "#E91E63"
)) +
labs(title = "GATA1 Knockout",
subtitle = "Cells deflected away from erythroid fate") +
theme_minimal() +
theme(panel.grid = element_blank(), legend.position = "none") +
coord_fixed()
p2 <- ggplot() +
geom_point(data = demo_data,
aes(x = UMAP_1, y = UMAP_2, color = cell_type),
alpha = 0.3, size = 0.8) +
geom_segment(data = grid,
aes(x = x, y = y, xend = x + dx_pu1ko, yend = y + dy_pu1ko),
arrow = arrow(length = unit(0.1, "cm")),
color = "black", alpha = 0.7) +
scale_color_manual(values = c(
"HSC" = "#9C27B0", "MPP" = "#7B1FA2", "CMP" = "#1976D2",
"GMP" = "#0288D1", "MEP" = "#0097A7", "Monocyte" = "#388E3C",
"Neutrophil" = "#689F38", "Erythrocyte" = "#F44336", "Megakaryocyte" = "#E91E63"
)) +
labs(title = "PU.1 (SPI1) Knockout",
subtitle = "Cells deflected away from myeloid fate") +
theme_minimal() +
theme(panel.grid = element_blank(), legend.position = "none") +
coord_fixed()
if (requireNamespace("patchwork", quietly = TRUE)) {
library(patchwork)
p1 + p2 + plot_annotation(
title = "In Silico TF Perturbation Simulations",
theme = theme(plot.title = element_text(face = "bold", size = 16))
)
} else {
print(p1)
}
Step 5: Analyze Fate Changes
# Simulate fate probability changes
perturbations <- c("Control", "GATA1 KO", "PU.1 KO", "CEBPA KO")
fates <- c("Erythrocyte", "Monocyte", "Neutrophil")
fate_probs <- data.frame(
perturbation = rep(perturbations, each = 3),
fate = rep(fates, 4),
probability = c(
0.35, 0.35, 0.30, # Control
0.10, 0.50, 0.40, # GATA1 KO (reduced erythroid)
0.55, 0.10, 0.35, # PU.1 KO (reduced myeloid)
0.40, 0.35, 0.25 # CEBPA KO (reduced neutrophil)
)
)
fate_probs$perturbation <- factor(fate_probs$perturbation, levels = perturbations)
ggplot(fate_probs, aes(x = perturbation, y = probability, fill = fate)) +
geom_col(position = "dodge", width = 0.7, color = "black") +
scale_fill_manual(values = c(
"Erythrocyte" = "#F44336",
"Monocyte" = "#388E3C",
"Neutrophil" = "#689F38"
)) +
labs(
title = "Predicted Cell Fate Changes Following TF Perturbation",
subtitle = "Starting from CMP population",
x = "Perturbation",
y = "Fate Probability",
fill = "Cell Fate"
) +
theme_bw() +
theme(
plot.title = element_text(face = "bold", size = 14),
axis.text.x = element_text(angle = 30, hjust = 1)
) +
geom_hline(yintercept = 1/3, linetype = "dashed", color = "gray50")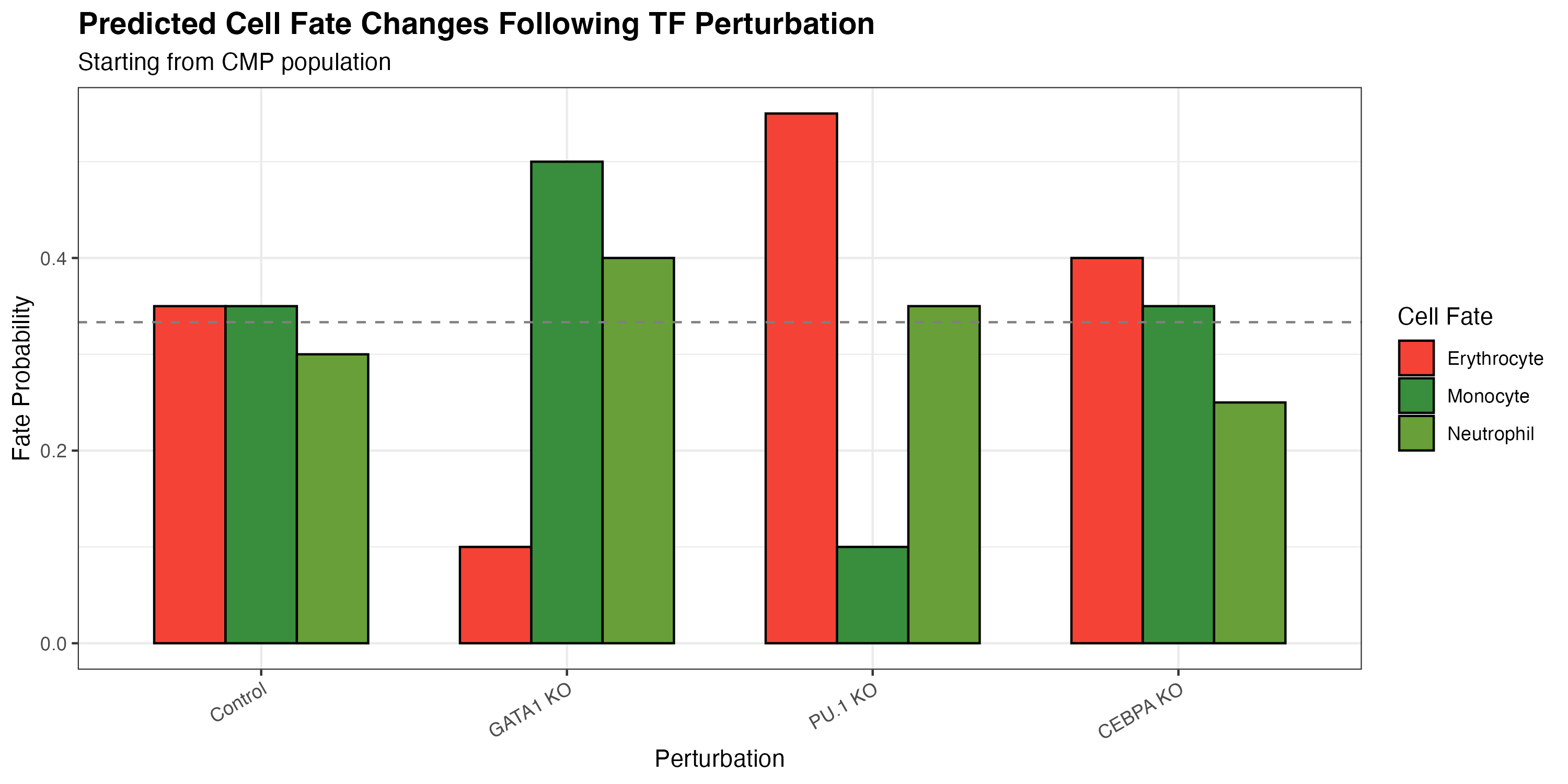
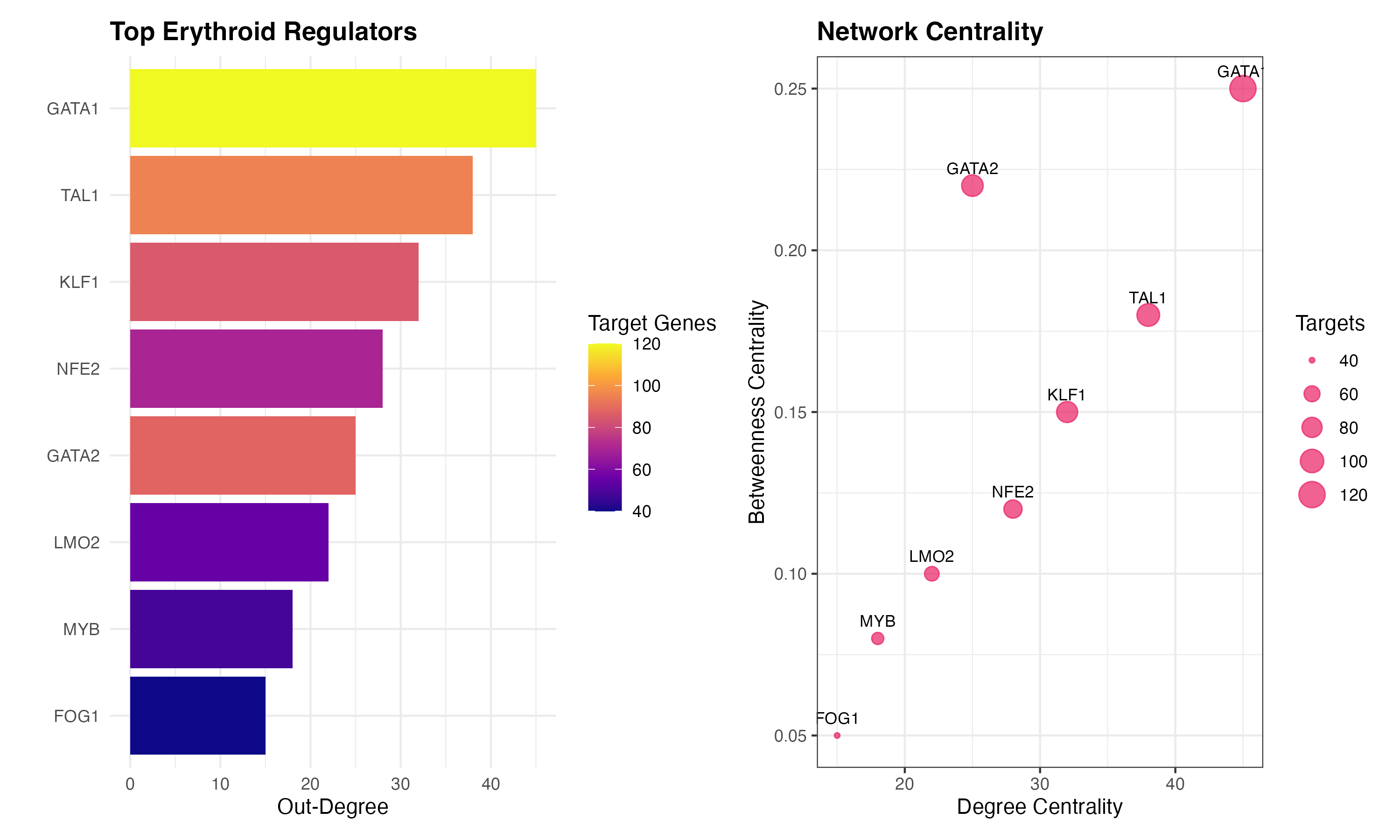
Step 6: Network Analysis
# Top regulators in erythroid lineage
erythroid_regulators <- data.frame(
TF = c("GATA1", "TAL1", "KLF1", "NFE2", "GATA2", "LMO2", "MYB", "FOG1"),
degree = c(45, 38, 32, 28, 25, 22, 18, 15),
betweenness = c(0.25, 0.18, 0.15, 0.12, 0.22, 0.10, 0.08, 0.05),
targets = c(120, 95, 85, 70, 88, 55, 48, 40)
)
# Order by degree
erythroid_regulators$TF <- factor(erythroid_regulators$TF,
levels = erythroid_regulators$TF[order(erythroid_regulators$degree)])
p1 <- ggplot(erythroid_regulators, aes(x = degree, y = TF, fill = targets)) +
geom_col() +
scale_fill_viridis_c(option = "plasma") +
labs(
title = "Top Erythroid Regulators",
x = "Out-Degree",
y = "",
fill = "Target Genes"
) +
theme_minimal() +
theme(plot.title = element_text(face = "bold"))
# Hub genes scatter
p2 <- ggplot(erythroid_regulators, aes(x = degree, y = betweenness,
size = targets, label = TF)) +
geom_point(alpha = 0.7, color = "#E91E63") +
geom_text(vjust = -1, size = 3) +
labs(
title = "Network Centrality",
x = "Degree Centrality",
y = "Betweenness Centrality",
size = "Targets"
) +
theme_bw() +
theme(plot.title = element_text(face = "bold"))
if (requireNamespace("patchwork", quietly = TRUE)) {
p1 + p2
} else {
print(p1)
}
Results Interpretation
GATA1 Knockout
- Observed: Reduced erythrocyte and megakaryocyte differentiation
- Compensated by: Increased myeloid cell production
- Biological relevance: Consistent with GATA1 knockout mouse phenotypes
Code for Full Analysis
library(CellOracleR)
library(Seurat)
# 1. Load data
seurat_obj <- readRDS("hematopoiesis_seurat.rds")
# 2. Create Oracle
oracle <- create_oracle(
seurat_obj,
cluster_col = "cell_type",
embedding_name = "umap"
)
# 3. Load base GRN
base_grn <- load_base_grn("hematopoiesis_base_grn.csv")
oracle$import_TF_data(TFdict = base_grn)
# 4. Fit GRN
oracle$fit_grn_for_simulation(
GRN_unit = "cluster",
alpha = 10,
bagging_number = 200
)
# 5. Simulate GATA1 knockout
oracle$simulate_perturbation(
perturb_condition = create_perturb_condition(
genes = "GATA1",
expression = 0 # knockout
),
n_propagation = 3
)
# 6. Calculate transition probabilities
oracle$estimate_transition_prob()
oracle$calculate_embedding_shift()
oracle$calculate_grid_arrows()
# 7. Visualize results
plot_simulation_flow(oracle, scale_factor = 1.5)
# 8. Save results
save_oracle(oracle, "gata1_ko_oracle.qs")Conclusions
CellOracleR enables systematic exploration of transcription factor function in cell fate decisions:
- Identify key regulators through network analysis
- Predict perturbation effects through GRN simulation
- Understand lineage relationships through trajectory analysis
- Generate testable hypotheses for experimental validation
References
Orkin, S.H. & Zon, L.I. (2008). Hematopoiesis: an evolving paradigm for stem cell biology. Cell.
Iwasaki, H. & Akashi, K. (2007). Myeloid lineage commitment from the hematopoietic stem cell. Immunity.
Pevny, L. et al. (1991). Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature.
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] patchwork_1.3.2 Matrix_1.7-4 ggplot2_4.0.1
#>
#> loaded via a namespace (and not attached):
#> [1] gtable_0.3.6 jsonlite_2.0.0 dplyr_1.1.4 compiler_4.4.0
#> [5] tidyselect_1.2.1 dichromat_2.0-0.1 jquerylib_0.1.4 systemfonts_1.3.1
#> [9] scales_1.4.0 textshaping_1.0.4 yaml_2.3.12 fastmap_1.2.0
#> [13] lattice_0.22-7 R6_2.6.1 labeling_0.4.3 generics_0.1.4
#> [17] knitr_1.51 htmlwidgets_1.6.4 tibble_3.3.1 desc_1.4.3
#> [21] bslib_0.9.0 pillar_1.11.1 RColorBrewer_1.1-3 rlang_1.1.7
#> [25] cachem_1.1.0 xfun_0.56 fs_1.6.6 sass_0.4.10
#> [29] S7_0.2.1 otel_0.2.0 viridisLite_0.4.2 cli_3.6.5
#> [33] pkgdown_2.1.3 withr_3.0.2 magrittr_2.0.4 digest_0.6.39
#> [37] grid_4.4.0 lifecycle_1.0.5 vctrs_0.7.1 evaluate_1.0.5
#> [41] glue_1.8.0 farver_2.1.2 ragg_1.5.0 rmarkdown_2.30
#> [45] tools_4.4.0 pkgconfig_2.0.3 htmltools_0.5.9