Algorithm Principles
Mathematical Foundations of CellOracleR
Zaoqu Liu
2026-01-25
Source:vignettes/algorithm-principles.Rmd
algorithm-principles.RmdIntroduction
CellOracleR implements a mathematical framework for predicting cell state transitions following transcription factor (TF) perturbations. This vignette describes the core algorithms underlying the package.
1. Gene Regulatory Network (GRN) Inference
1.1 Mathematical Formulation
For each target gene , we model its expression as a linear function of its regulators:
where:
- is the expression of target gene
- is the set of regulators for gene
- is the regulatory coefficient from regulator to target
- is the residual error
1.2 Ridge Regression
We use Ridge regression (L2 regularization) to estimate coefficients:
The closed-form solution is:
Why Ridge Regression?
- Prevents overfitting: Regularization shrinks coefficients toward zero
- Handles multicollinearity: Correlated regulators don’t cause instability
- Numerical stability: Always has a unique solution
# Ridge regression in CellOracleR
coef_matrix <- fit_grn_coef_matrix(
gem = expression_matrix,
TFdict = tf_target_dict,
alpha = 10 # regularization strength
)1.3 Bootstrap Aggregation (Bagging)
To improve robustness, we employ bagging:
- Sample 80% of cells (without replacement)
- Fit Ridge regression on the subsample
- Repeat times (typically 200)
- Use median of coefficients as final estimate
Advantages:
- Reduces variance of coefficient estimates
- More robust to outlier cells
- Provides coefficient stability measures
2. Signal Propagation Simulation
2.1 Perturbation Model
Given a perturbation condition (e.g., TF knockout), we simulate the downstream effects:
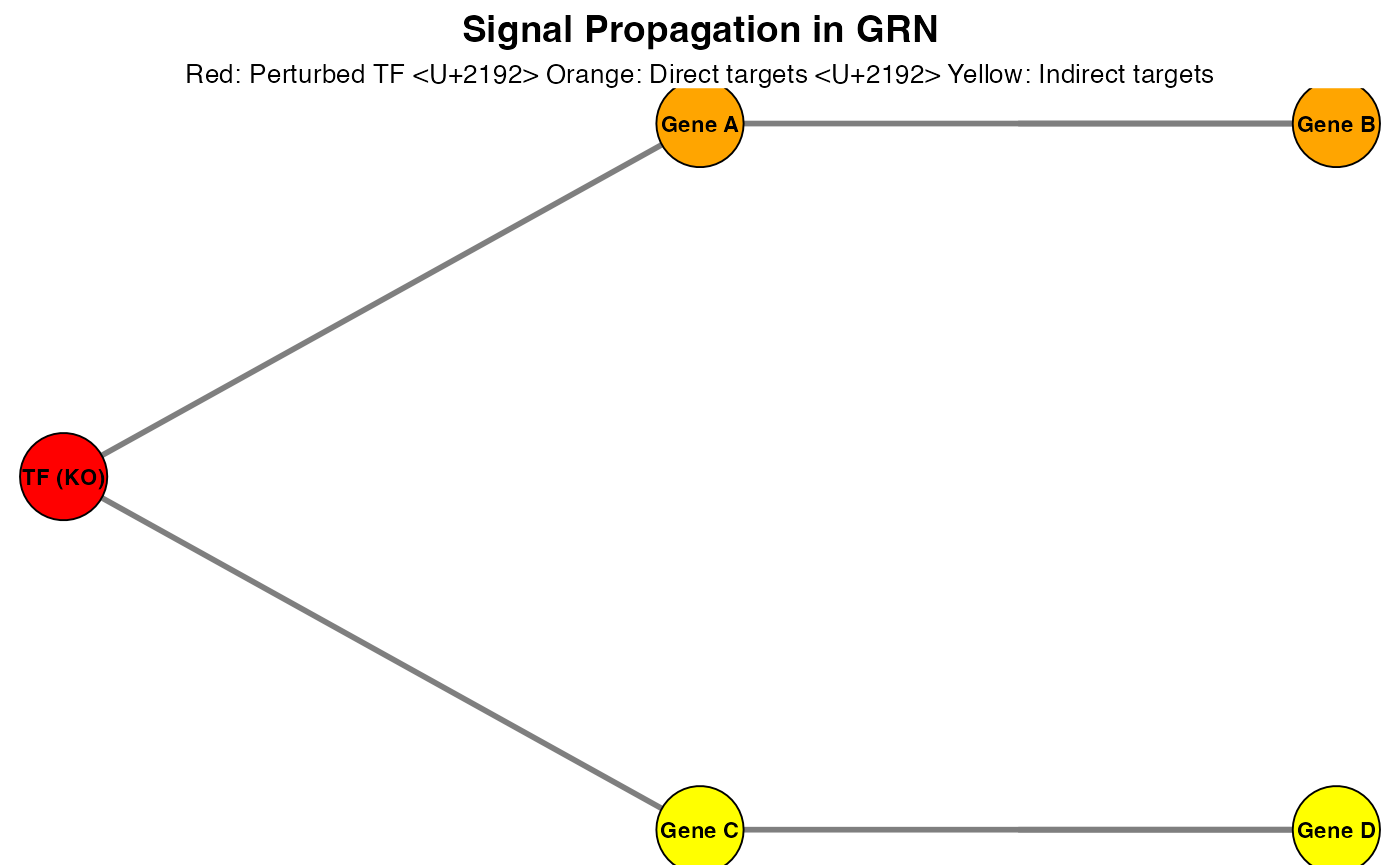
3. Transition Probability Estimation
3.1 Correlation-Based Approach
We estimate the probability of cell transitioning to cell based on:
This measures how well the predicted expression change aligns with the direction toward cell .
4. Markov Chain Simulation
Summary
CellOracleR combines:
- Ridge regression for robust GRN inference
-
Signal propagation for perturbation
simulation
- Correlation-based transition probabilities for fate prediction
- Markov chains for trajectory modeling
- Graph theory for network analysis
These mathematical foundations enable accurate prediction of cellular responses to genetic perturbations.
References
Kamimoto, K., et al. (2023). CellOracle: Dissecting cell identity via network inference and in silico gene perturbation. Molecular Systems Biology.
Hoerl, A.E. & Kennard, R.W. (1970). Ridge Regression: Biased Estimation for Nonorthogonal Problems. Technometrics.
Breiman, L. (1996). Bagging Predictors. Machine Learning.
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggplot2_4.0.1
#>
#> loaded via a namespace (and not attached):
#> [1] gtable_0.3.6 jsonlite_2.0.0 dplyr_1.1.4 compiler_4.4.0
#> [5] tidyselect_1.2.1 dichromat_2.0-0.1 jquerylib_0.1.4 systemfonts_1.3.1
#> [9] scales_1.4.0 textshaping_1.0.4 yaml_2.3.12 fastmap_1.2.0
#> [13] R6_2.6.1 labeling_0.4.3 generics_0.1.4 knitr_1.51
#> [17] htmlwidgets_1.6.4 tibble_3.3.1 desc_1.4.3 bslib_0.9.0
#> [21] pillar_1.11.1 RColorBrewer_1.1-3 rlang_1.1.7 cachem_1.1.0
#> [25] xfun_0.56 fs_1.6.6 sass_0.4.10 S7_0.2.1
#> [29] otel_0.2.0 cli_3.6.5 pkgdown_2.1.3 withr_3.0.2
#> [33] magrittr_2.0.4 digest_0.6.39 grid_4.4.0 lifecycle_1.0.5
#> [37] vctrs_0.7.1 evaluate_1.0.5 glue_1.8.0 farver_2.1.2
#> [41] ragg_1.5.0 rmarkdown_2.30 tools_4.4.0 pkgconfig_2.0.3
#> [45] htmltools_0.5.9