GRN Inference Details
Building Gene Regulatory Networks from Single-Cell Data
Zaoqu Liu
2026-01-25
Source:vignettes/grn-inference.Rmd
grn-inference.RmdOverview
Gene Regulatory Network (GRN) inference is the foundation of CellOracleR analysis. This vignette provides detailed documentation of the GRN inference pipeline.
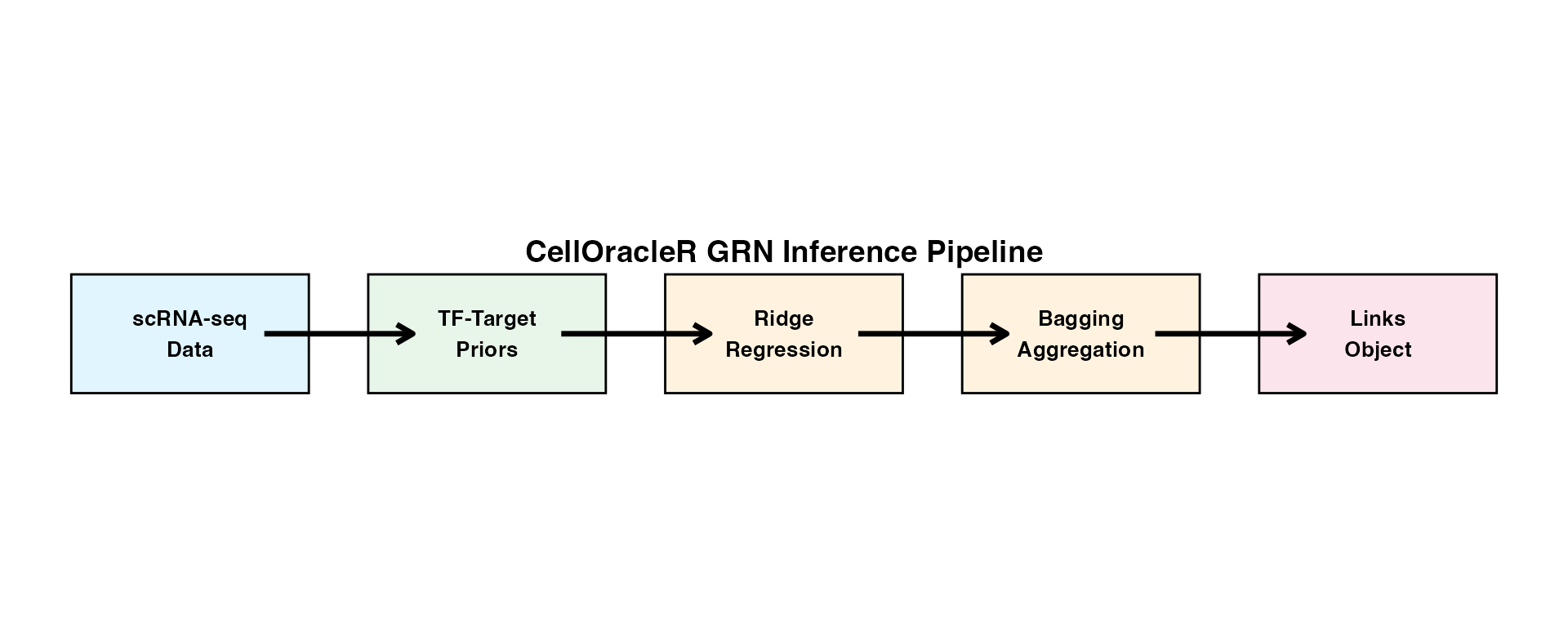
Step 1: Input Data Preparation
Expression Matrix
CellOracleR requires a normalized expression matrix (genes × cells):
# From Seurat object
library(Seurat)
library(CellOracleR)
# Create Oracle object
oracle <- create_oracle(
seurat_obj,
cluster_col = "seurat_clusters",
embedding_name = "umap"
)
# Extract expression data
expr_matrix <- oracle$get_expression()
dim(expr_matrix) # genes × cellsRecommendations:
- Use normalized data (log-transformed)
- Filter low-quality cells and genes
- 2,000-5,000 highly variable genes typically sufficient
TF-Target Prior Dictionary
The TF-target dictionary specifies which TFs can potentially regulate each gene:
# Structure: list with gene names as keys
# Each element contains a character vector of potential TF regulators
TFdict <- list(
Gene1 = c("TF_A", "TF_B", "TF_C"),
Gene2 = c("TF_A", "TF_D"),
Gene3 = c("TF_B", "TF_C", "TF_D", "TF_E")
)Sources for TF-target priors:
- Motif analysis (recommended): Scan promoter sequences for TF binding motifs
- ChIP-seq data: Experimentally determined TF binding sites
- Literature curation: Known regulatory relationships
- ATAC-seq + Cicero: Accessibility-based predictions
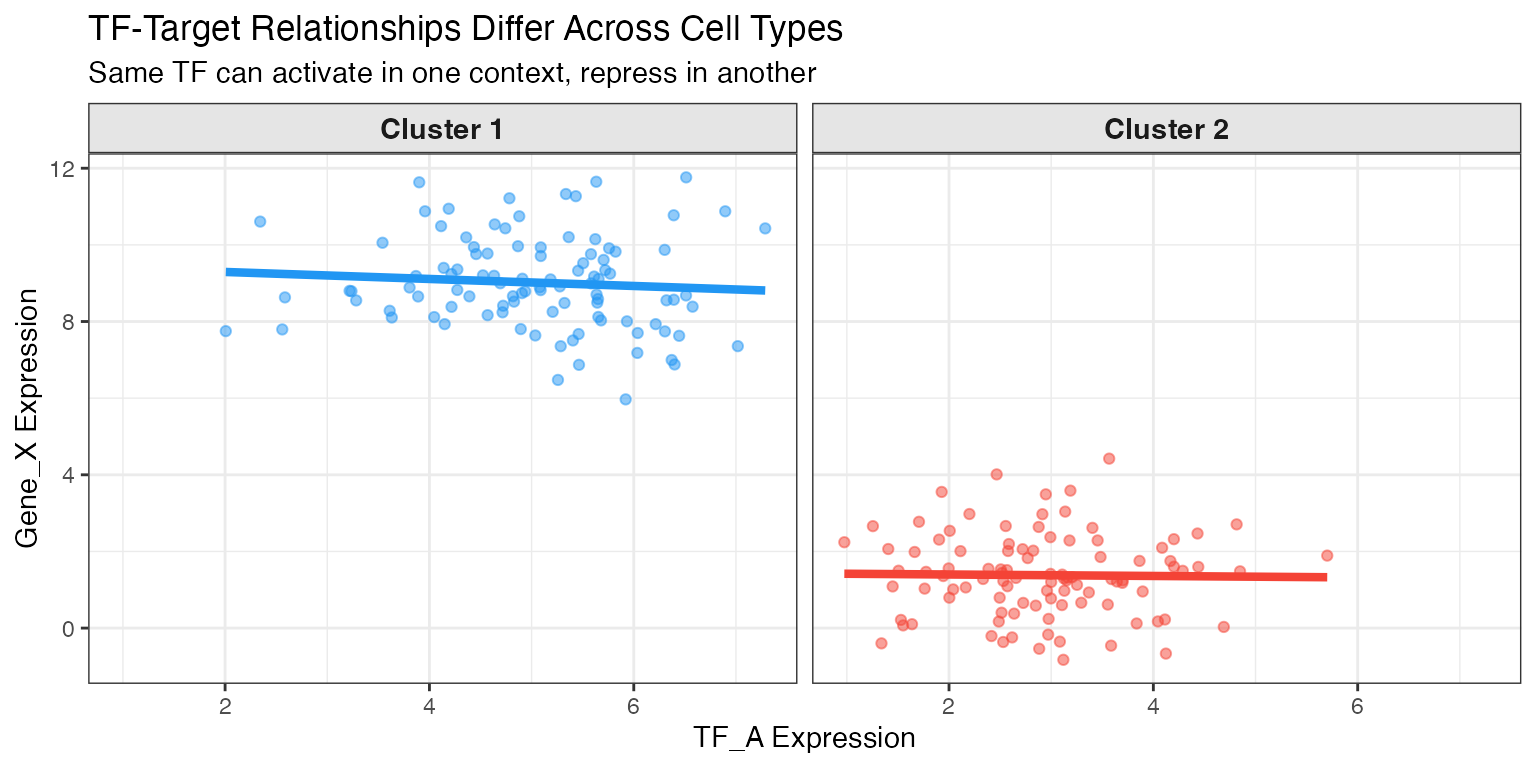
Step 2: Cluster-Specific GRN Fitting
GRNs are fitted separately for each cell cluster to capture cell-type-specific regulatory logic.
Step 3: Ridge Regression Details
Regularization Parameter (α)
The regularization parameter controls the bias-variance trade-off:
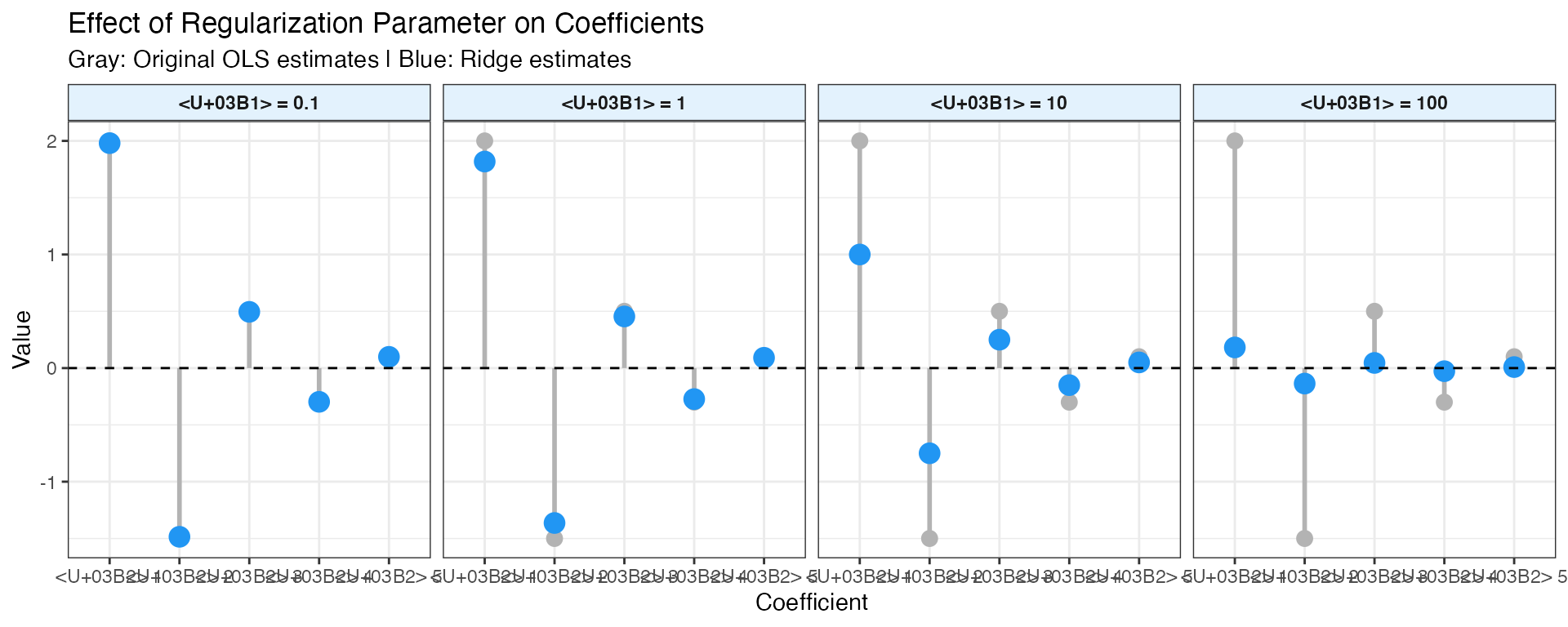
Guidelines for choosing α:
| α value | Effect | Use case |
|---|---|---|
| 0.1-1 | Minimal shrinkage | High-quality data, few predictors |
| 1-10 | Moderate shrinkage | Standard scRNA-seq (recommended) |
| 10-100 | Strong shrinkage | Noisy data, many predictors |
| >100 | Heavy shrinkage | Highly correlated predictors |
Default: α = 10 - provides good balance for typical scRNA-seq data.
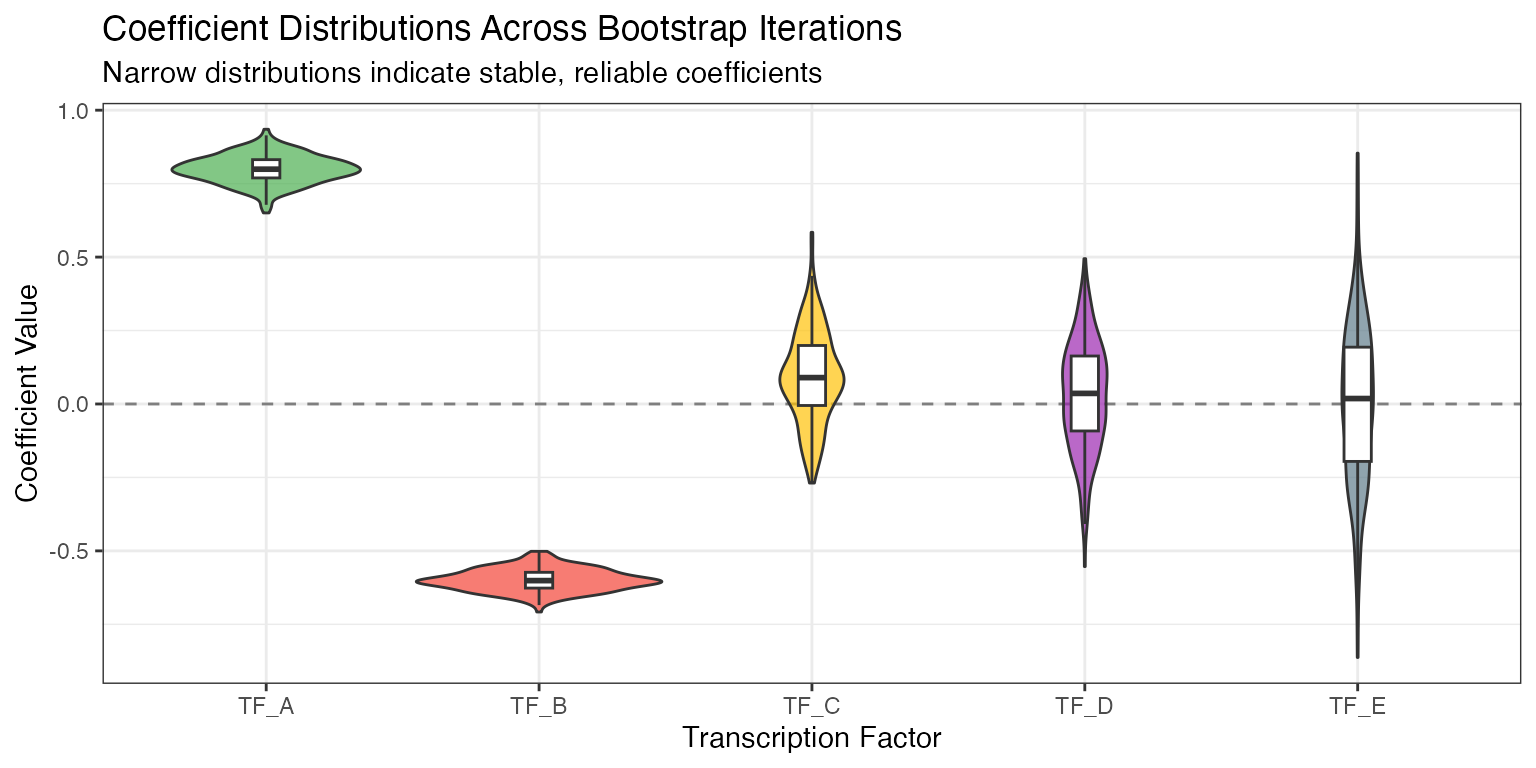
Step 4: Bootstrap Aggregation
Algorithm
# Pseudocode for bagging
bagging_coefficients <- function(X, y, n_bootstrap = 200, sample_ratio = 0.8) {
n_cells <- ncol(X)
n_sample <- floor(n_cells * sample_ratio)
coef_list <- list()
for (b in 1:n_bootstrap) {
# Random subsample without replacement
idx <- sample(n_cells, n_sample, replace = FALSE)
# Fit Ridge regression
coef_list[[b]] <- ridge_fit(X[, idx], y[idx], alpha = 10)
}
# Aggregate: use median (robust to outliers)
final_coef <- apply(do.call(cbind, coef_list), 1, median)
return(final_coef)
}
Step 5: Links Object
The Links class stores and analyzes the fitted GRN:
# Get Links object from Oracle
links <- oracle$get_links(
cluster_name_for_GRN_unit = "Cluster1"
)
# Explore the network
links$filter(
threshold_p = 0.001, # p-value threshold
threshold_coef = 0.1 # coefficient magnitude threshold
)
# Calculate network metrics
links$get_network_score()
# Export for visualization
links$get_igraph()Performance Considerations
Computational Complexity
| Component | Time Complexity | Memory |
|---|---|---|
| Ridge regression | O(p³) per gene | O(p²) |
| Bagging (B iterations) | O(B × n × p³) | O(p²) |
| Full GRN | O(G × B × n × p³) | O(G × p) |
Where: G = genes, n = cells, p = max TFs per gene, B = bootstrap iterations
Best Practices
1. Data Quality
# Filter before GRN inference
seurat_obj <- subset(seurat_obj,
nFeature_RNA > 500 &
nCount_RNA > 1000 &
percent.mt < 20)Summary
The CellOracleR GRN inference pipeline:
- Prepares expression data and TF-target priors
- Fits cluster-specific Ridge regression models
- Stabilizes estimates through bootstrap aggregation
- Stores results in the Links class for downstream analysis
This produces robust, interpretable gene regulatory networks suitable for perturbation simulation.
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] Matrix_1.7-4 ggplot2_4.0.1
#>
#> loaded via a namespace (and not attached):
#> [1] gtable_0.3.6 jsonlite_2.0.0 dplyr_1.1.4 compiler_4.4.0
#> [5] tidyselect_1.2.1 dichromat_2.0-0.1 jquerylib_0.1.4 splines_4.4.0
#> [9] systemfonts_1.3.1 scales_1.4.0 textshaping_1.0.4 yaml_2.3.12
#> [13] fastmap_1.2.0 lattice_0.22-7 R6_2.6.1 labeling_0.4.3
#> [17] generics_0.1.4 knitr_1.51 htmlwidgets_1.6.4 tibble_3.3.1
#> [21] desc_1.4.3 bslib_0.9.0 pillar_1.11.1 RColorBrewer_1.1-3
#> [25] rlang_1.1.7 cachem_1.1.0 xfun_0.56 fs_1.6.6
#> [29] sass_0.4.10 S7_0.2.1 otel_0.2.0 cli_3.6.5
#> [33] mgcv_1.9-3 pkgdown_2.1.3 withr_3.0.2 magrittr_2.0.4
#> [37] digest_0.6.39 grid_4.4.0 nlme_3.1-168 lifecycle_1.0.5
#> [41] vctrs_0.7.1 evaluate_1.0.5 glue_1.8.0 farver_2.1.2
#> [45] ragg_1.5.0 rmarkdown_2.30 tools_4.4.0 pkgconfig_2.0.3
#> [49] htmltools_0.5.9