Visualization Gallery
Comprehensive Guide to CellOracleR Plotting Functions
Zaoqu Liu
2026-01-25
Source:vignettes/visualization-gallery.Rmd
visualization-gallery.RmdIntroduction
CellOracleR provides a comprehensive suite of visualization functions built on ggplot2. This gallery demonstrates the available plot types and customization options.
Cell Embedding Visualizations
Basic Cluster Plot
Visualize cell clusters in embedding space:
# Generate demo data
set.seed(42)
n_cells <- 500
# Create clusters with different distributions
demo_embedding <- data.frame(
UMAP_1 = c(rnorm(200, -3, 0.8), rnorm(150, 2, 1), rnorm(150, 0, 0.6)),
UMAP_2 = c(rnorm(200, 0, 0.8), rnorm(150, 2, 0.9), rnorm(150, -2, 0.7)),
cluster = factor(c(rep("HSC", 200), rep("Monocyte", 150), rep("Erythroid", 150)))
)
# Cluster plot
ggplot(demo_embedding, aes(x = UMAP_1, y = UMAP_2, color = cluster)) +
geom_point(alpha = 0.6, size = 1.5) +
scale_color_manual(values = c("#E41A1C", "#377EB8", "#4DAF4A")) +
labs(
title = "Cell Clusters in UMAP Space",
subtitle = "CellOracleR plot_cluster()",
x = "UMAP 1",
y = "UMAP 2",
color = "Cell Type"
) +
theme_minimal() +
theme(
panel.grid = element_blank(),
legend.position = "right",
plot.title = element_text(face = "bold", size = 14)
) +
coord_fixed()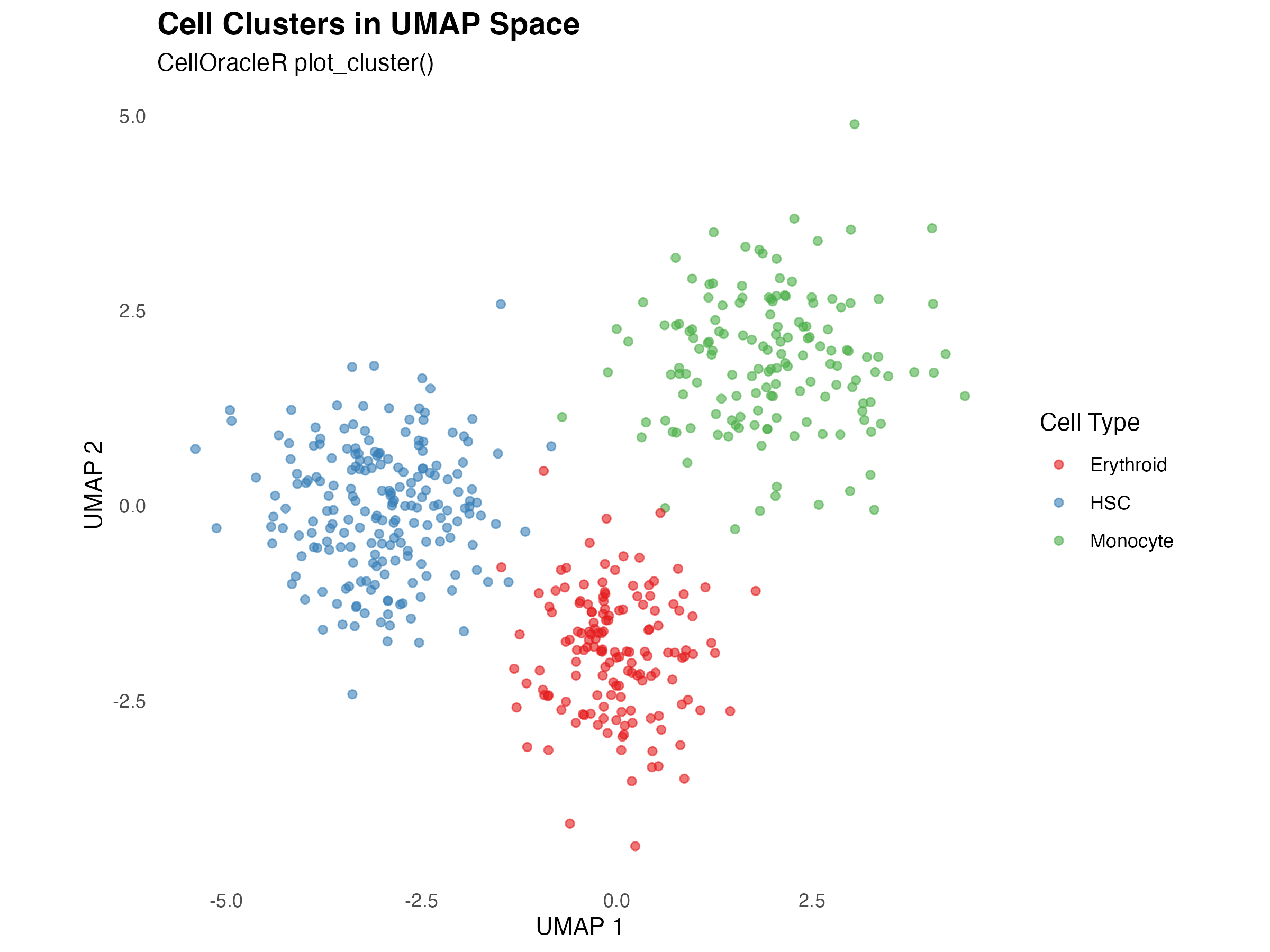
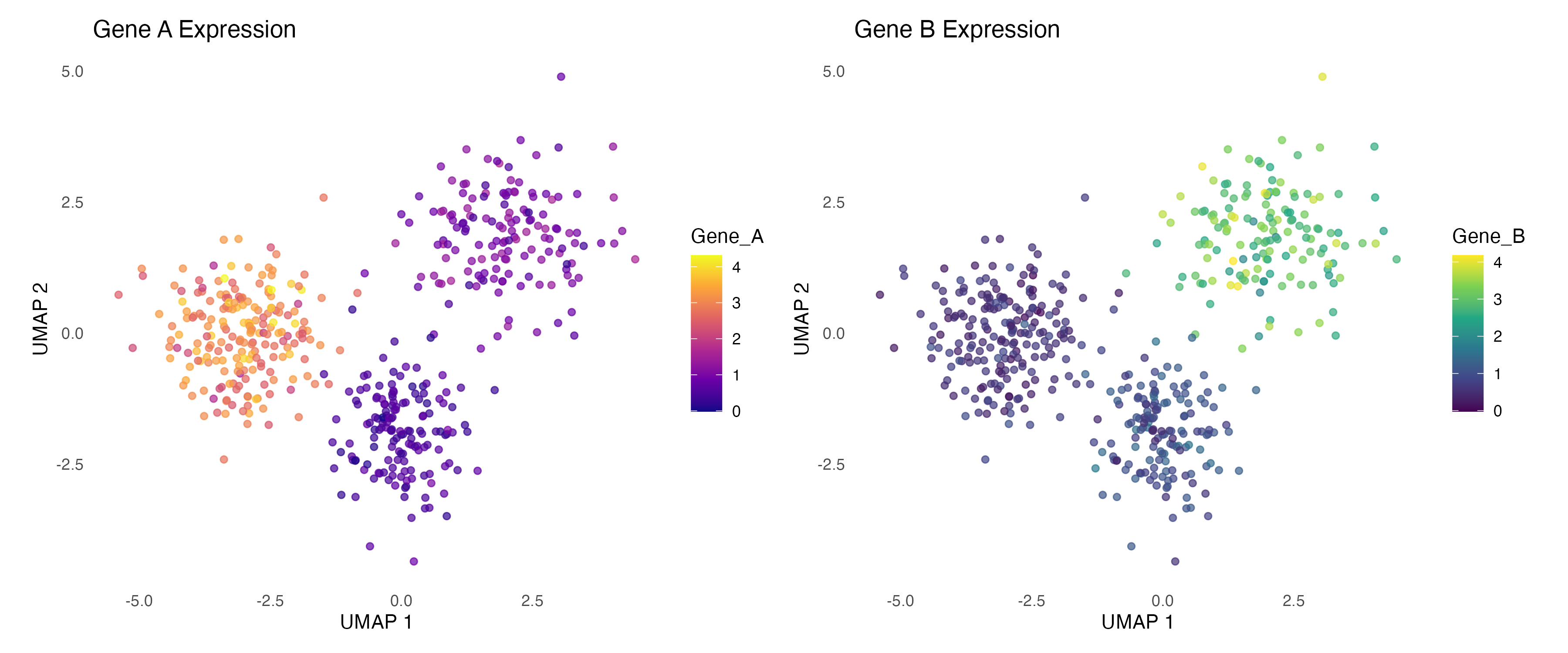
Gene Expression Overlay
Visualize gene expression on embedding:
# Add expression data
demo_embedding$Gene_A <- c(
rnorm(200, 3, 0.5), # High in HSC
rnorm(150, 1, 0.3), # Low in Monocyte
rnorm(150, 0.5, 0.2) # Very low in Erythroid
)
demo_embedding$Gene_B <- c(
rnorm(200, 0.5, 0.2), # Low in HSC
rnorm(150, 3, 0.5), # High in Monocyte
rnorm(150, 1, 0.3) # Medium in Erythroid
)
# Create side-by-side plots
p1 <- ggplot(demo_embedding, aes(x = UMAP_1, y = UMAP_2, color = Gene_A)) +
geom_point(alpha = 0.7, size = 1.5) +
scale_color_viridis_c(option = "plasma") +
labs(title = "Gene A Expression", x = "UMAP 1", y = "UMAP 2") +
theme_minimal() +
theme(panel.grid = element_blank()) +
coord_fixed()
p2 <- ggplot(demo_embedding, aes(x = UMAP_1, y = UMAP_2, color = Gene_B)) +
geom_point(alpha = 0.7, size = 1.5) +
scale_color_viridis_c(option = "viridis") +
labs(title = "Gene B Expression", x = "UMAP 1", y = "UMAP 2") +
theme_minimal() +
theme(panel.grid = element_blank()) +
coord_fixed()
if (requireNamespace("patchwork", quietly = TRUE)) {
library(patchwork)
p1 + p2
} else {
print(p1)
}
Simulation Flow Visualizations
Quiver Plot (Vector Field)
The quiver plot shows predicted cell movement directions:
# Create grid for quiver plot
grid_x <- seq(-5, 4, by = 1)
grid_y <- seq(-4, 4, by = 1)
grid_data <- expand.grid(x = grid_x, y = grid_y)
# Simulate flow vectors (pointing toward attractors)
attractor1 <- c(-3, 0)
attractor2 <- c(2, 2)
# Calculate vectors
grid_data$dx <- 0
grid_data$dy <- 0
for (i in 1:nrow(grid_data)) {
# Distance to attractors
d1 <- sqrt((grid_data$x[i] - attractor1[1])^2 + (grid_data$y[i] - attractor1[2])^2)
d2 <- sqrt((grid_data$x[i] - attractor2[1])^2 + (grid_data$y[i] - attractor2[2])^2)
# Weight by inverse distance
w1 <- 1 / (d1 + 0.5)^2
w2 <- 1 / (d2 + 0.5)^2
# Combined direction
grid_data$dx[i] <- w1 * (attractor1[1] - grid_data$x[i]) +
w2 * (attractor2[1] - grid_data$x[i])
grid_data$dy[i] <- w1 * (attractor1[2] - grid_data$y[i]) +
w2 * (attractor2[2] - grid_data$y[i])
# Normalize
mag <- sqrt(grid_data$dx[i]^2 + grid_data$dy[i]^2)
if (mag > 0) {
grid_data$dx[i] <- grid_data$dx[i] / mag * 0.4
grid_data$dy[i] <- grid_data$dy[i] / mag * 0.4
}
}
# Calculate magnitude for coloring
grid_data$magnitude <- sqrt(grid_data$dx^2 + grid_data$dy^2)
ggplot() +
geom_point(data = demo_embedding,
aes(x = UMAP_1, y = UMAP_2, color = cluster),
alpha = 0.3, size = 1) +
geom_segment(data = grid_data,
aes(x = x, y = y, xend = x + dx, yend = y + dy),
arrow = arrow(length = unit(0.15, "cm"), type = "closed"),
color = "black", size = 0.6) +
scale_color_manual(values = c("#E41A1C", "#377EB8", "#4DAF4A")) +
labs(
title = "Simulation Flow Field (Quiver Plot)",
subtitle = "Arrows indicate predicted cell movement direction",
x = "UMAP 1",
y = "UMAP 2"
) +
theme_minimal() +
theme(
panel.grid = element_blank(),
plot.title = element_text(face = "bold", size = 14)
) +
coord_fixed()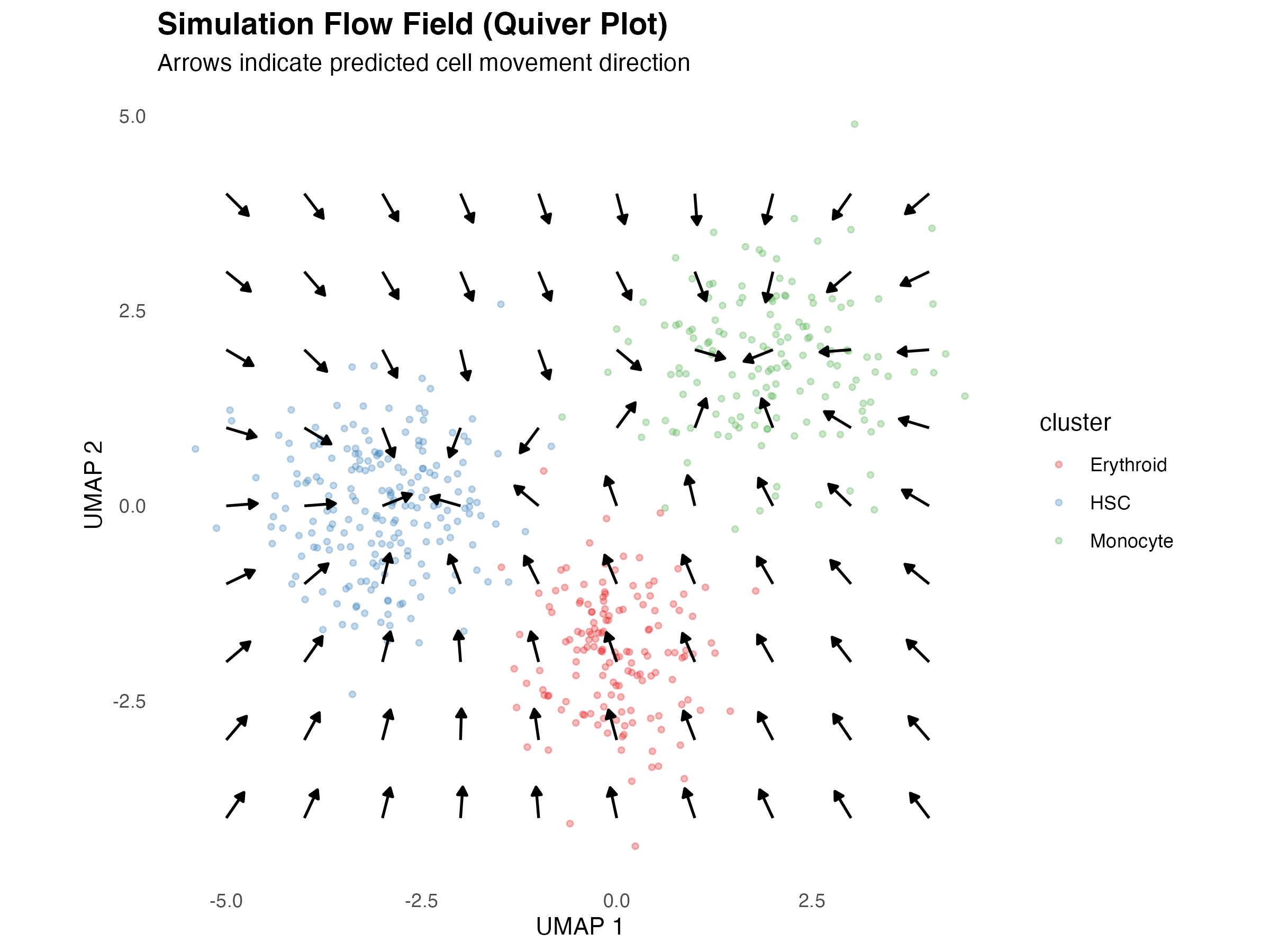
Streamline Plot
Smoother representation of flow patterns:
# Generate streamlines
set.seed(42)
n_streams <- 15
stream_length <- 50
streamlines <- list()
for (s in 1:n_streams) {
# Random starting points
start_x <- runif(1, -4, 3)
start_y <- runif(1, -3, 3)
stream <- data.frame(x = numeric(stream_length),
y = numeric(stream_length),
stream_id = s)
stream$x[1] <- start_x
stream$y[1] <- start_y
for (i in 2:stream_length) {
# Calculate direction
d1 <- sqrt((stream$x[i-1] - attractor1[1])^2 + (stream$y[i-1] - attractor1[2])^2)
d2 <- sqrt((stream$x[i-1] - attractor2[1])^2 + (stream$y[i-1] - attractor2[2])^2)
w1 <- 1 / (d1 + 0.5)^2
w2 <- 1 / (d2 + 0.5)^2
dx <- w1 * (attractor1[1] - stream$x[i-1]) + w2 * (attractor2[1] - stream$x[i-1])
dy <- w1 * (attractor1[2] - stream$y[i-1]) + w2 * (attractor2[2] - stream$y[i-1])
mag <- sqrt(dx^2 + dy^2)
if (mag > 0) {
stream$x[i] <- stream$x[i-1] + dx / mag * 0.15
stream$y[i] <- stream$y[i-1] + dy / mag * 0.15
} else {
stream$x[i] <- stream$x[i-1]
stream$y[i] <- stream$y[i-1]
}
}
stream$step <- 1:stream_length
streamlines[[s]] <- stream
}
stream_df <- do.call(rbind, streamlines)
ggplot() +
geom_point(data = demo_embedding,
aes(x = UMAP_1, y = UMAP_2, color = cluster),
alpha = 0.3, size = 1) +
geom_path(data = stream_df,
aes(x = x, y = y, group = stream_id, alpha = step),
color = "darkblue", size = 0.8) +
scale_color_manual(values = c("#E41A1C", "#377EB8", "#4DAF4A")) +
scale_alpha_continuous(range = c(0.2, 1), guide = "none") +
labs(
title = "Simulation Streamlines",
subtitle = "Continuous flow paths through the embedding",
x = "UMAP 1",
y = "UMAP 2"
) +
theme_minimal() +
theme(
panel.grid = element_blank(),
plot.title = element_text(face = "bold", size = 14)
) +
coord_fixed()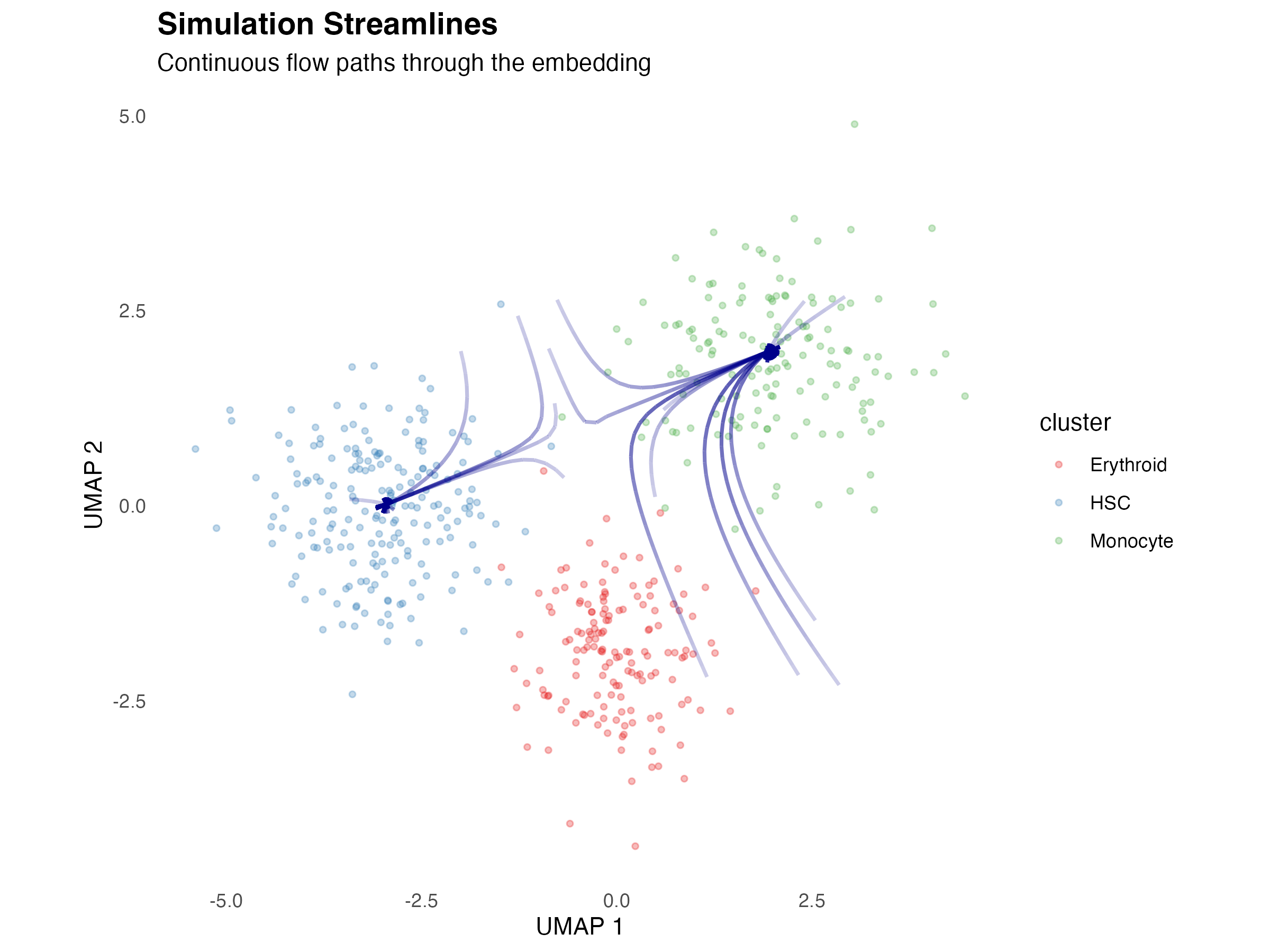
Network Visualizations
Network Graph
Visualize gene regulatory networks:
# Create demo network data
set.seed(42)
nodes <- data.frame(
name = c("TF1", "TF2", "TF3", "Gene_A", "Gene_B", "Gene_C", "Gene_D", "Gene_E"),
type = c(rep("TF", 3), rep("Target", 5)),
degree = c(4, 3, 2, 2, 2, 1, 1, 1)
)
edges <- data.frame(
from = c("TF1", "TF1", "TF1", "TF1", "TF2", "TF2", "TF2", "TF3", "TF3"),
to = c("Gene_A", "Gene_B", "Gene_C", "Gene_D", "Gene_A", "Gene_B", "Gene_E", "Gene_C", "Gene_D"),
weight = c(0.8, 0.6, 0.5, 0.3, 0.7, 0.4, 0.5, 0.6, 0.4)
)
# Create layout (circular for TFs, radial for targets)
node_pos <- data.frame(
name = nodes$name,
x = c(-2, 0, 2, -2, 0, 2, -1, 1),
y = c(2, 2, 2, -1, -1, -1, -2, -2)
)
# Merge positions
edges_plot <- merge(edges, node_pos, by.x = "from", by.y = "name")
names(edges_plot)[4:5] <- c("x_from", "y_from")
edges_plot <- merge(edges_plot, node_pos, by.x = "to", by.y = "name")
names(edges_plot)[6:7] <- c("x_to", "y_to")
nodes <- merge(nodes, node_pos, by = "name")
ggplot() +
geom_segment(data = edges_plot,
aes(x = x_from, y = y_from, xend = x_to, yend = y_to,
alpha = weight),
arrow = arrow(length = unit(0.25, "cm"), type = "closed"),
color = "gray40", size = 1) +
geom_point(data = nodes,
aes(x = x, y = y, fill = type, size = degree),
shape = 21, color = "black") +
geom_text(data = nodes,
aes(x = x, y = y, label = name),
vjust = -1.5, size = 3.5, fontface = "bold") +
scale_fill_manual(values = c("TF" = "#FF7043", "Target" = "#42A5F5")) +
scale_size_continuous(range = c(6, 12)) +
scale_alpha_continuous(range = c(0.3, 1)) +
labs(
title = "Gene Regulatory Network",
subtitle = "TFs (orange) regulate target genes (blue)",
fill = "Node Type",
size = "Degree"
) +
theme_void() +
theme(
plot.title = element_text(face = "bold", size = 14, hjust = 0.5),
plot.subtitle = element_text(hjust = 0.5),
legend.position = "bottom"
) +
guides(alpha = "none")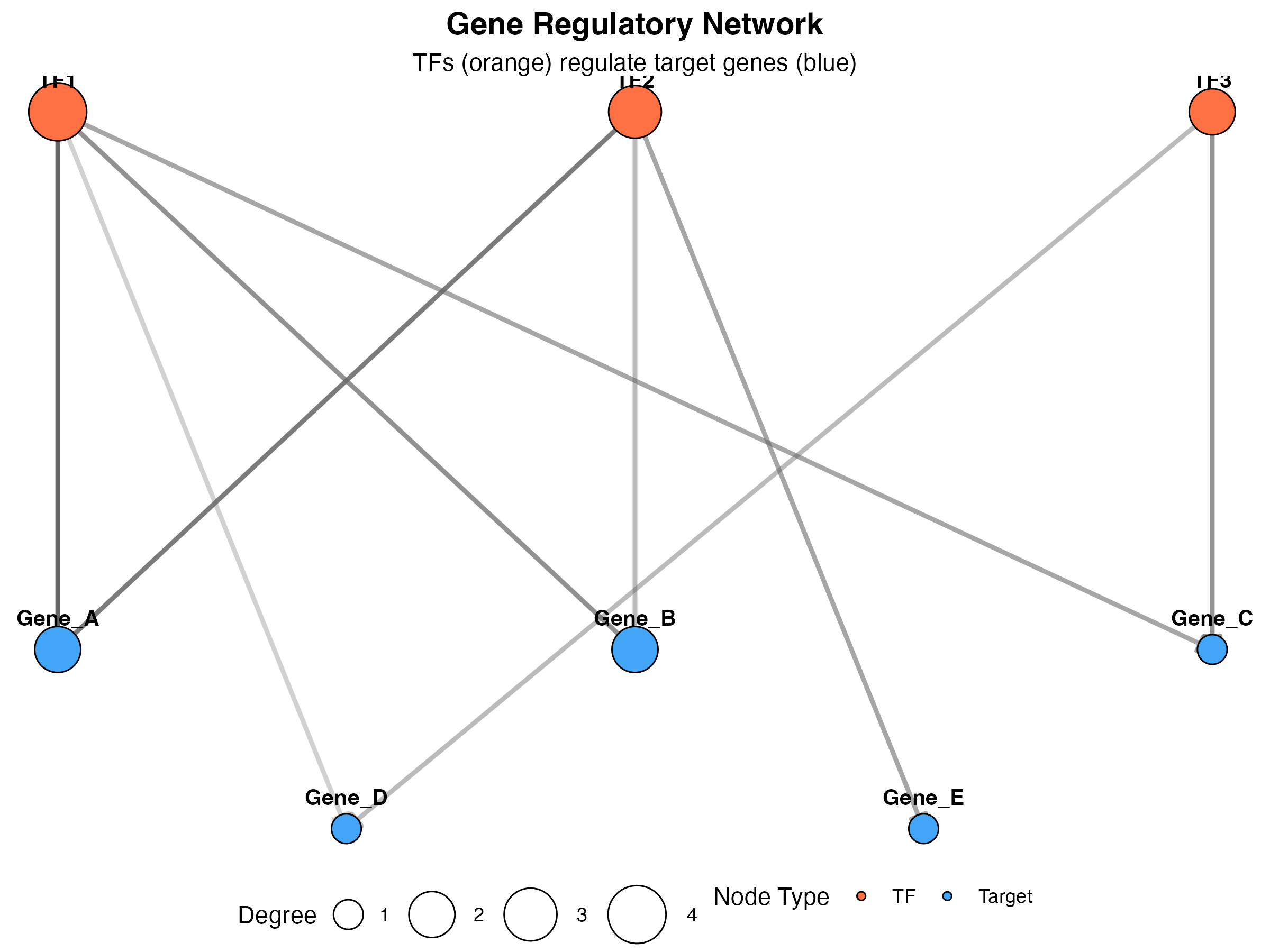
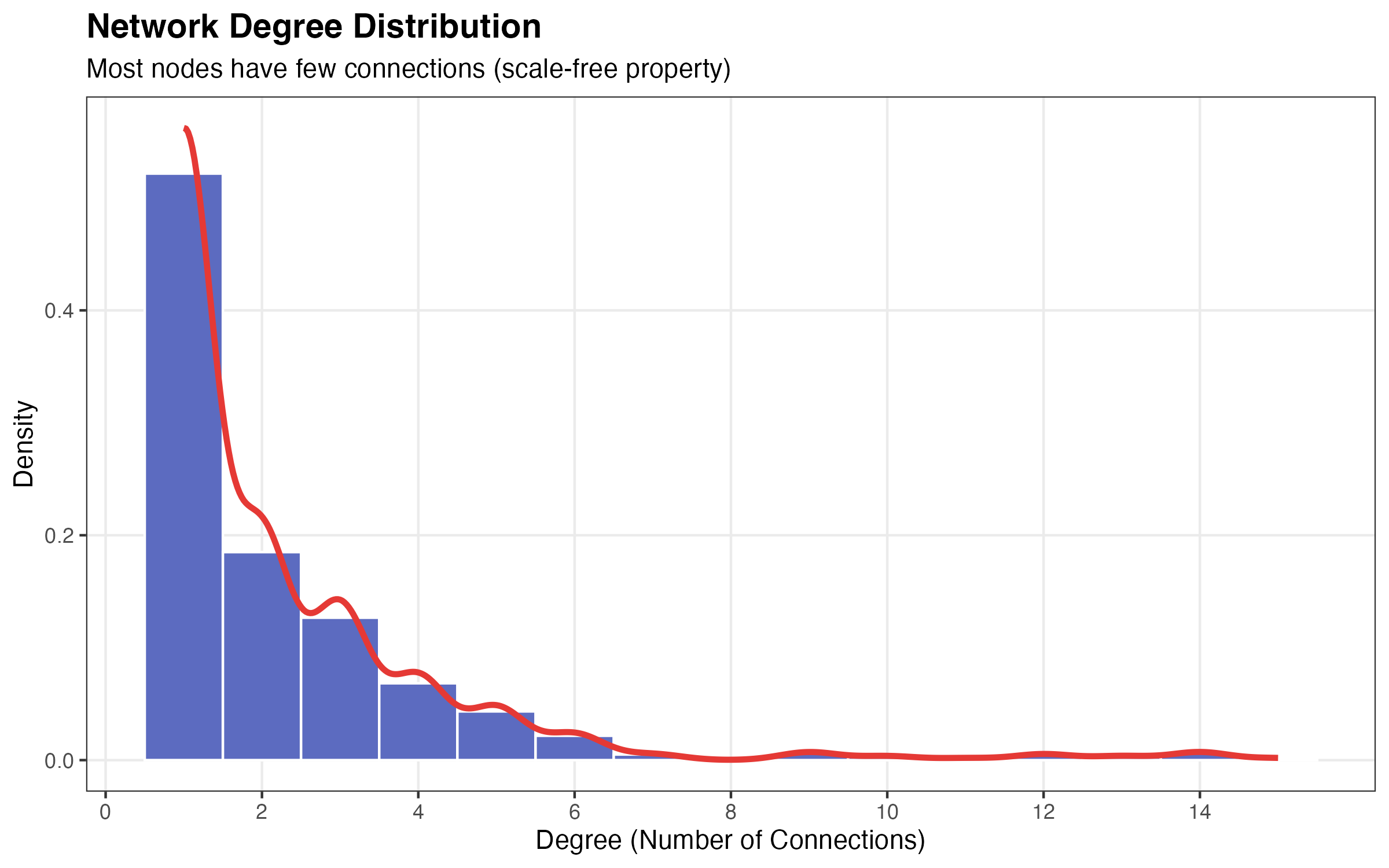
Degree Distribution
# Generate realistic degree distribution (power-law like)
set.seed(42)
degrees <- c(
sample(1:3, 500, replace = TRUE, prob = c(0.6, 0.25, 0.15)),
sample(4:6, 80, replace = TRUE, prob = c(0.5, 0.3, 0.2)),
sample(7:15, 20, replace = TRUE)
)
degree_df <- data.frame(degree = degrees)
ggplot(degree_df, aes(x = degree)) +
geom_histogram(aes(y = after_stat(density)),
binwidth = 1, fill = "#5C6BC0", color = "white") +
geom_density(color = "#E53935", size = 1.2) +
scale_x_continuous(breaks = seq(0, 15, 2)) +
labs(
title = "Network Degree Distribution",
subtitle = "Most nodes have few connections (scale-free property)",
x = "Degree (Number of Connections)",
y = "Density"
) +
theme_bw() +
theme(
plot.title = element_text(face = "bold", size = 14),
panel.grid.minor = element_blank()
)
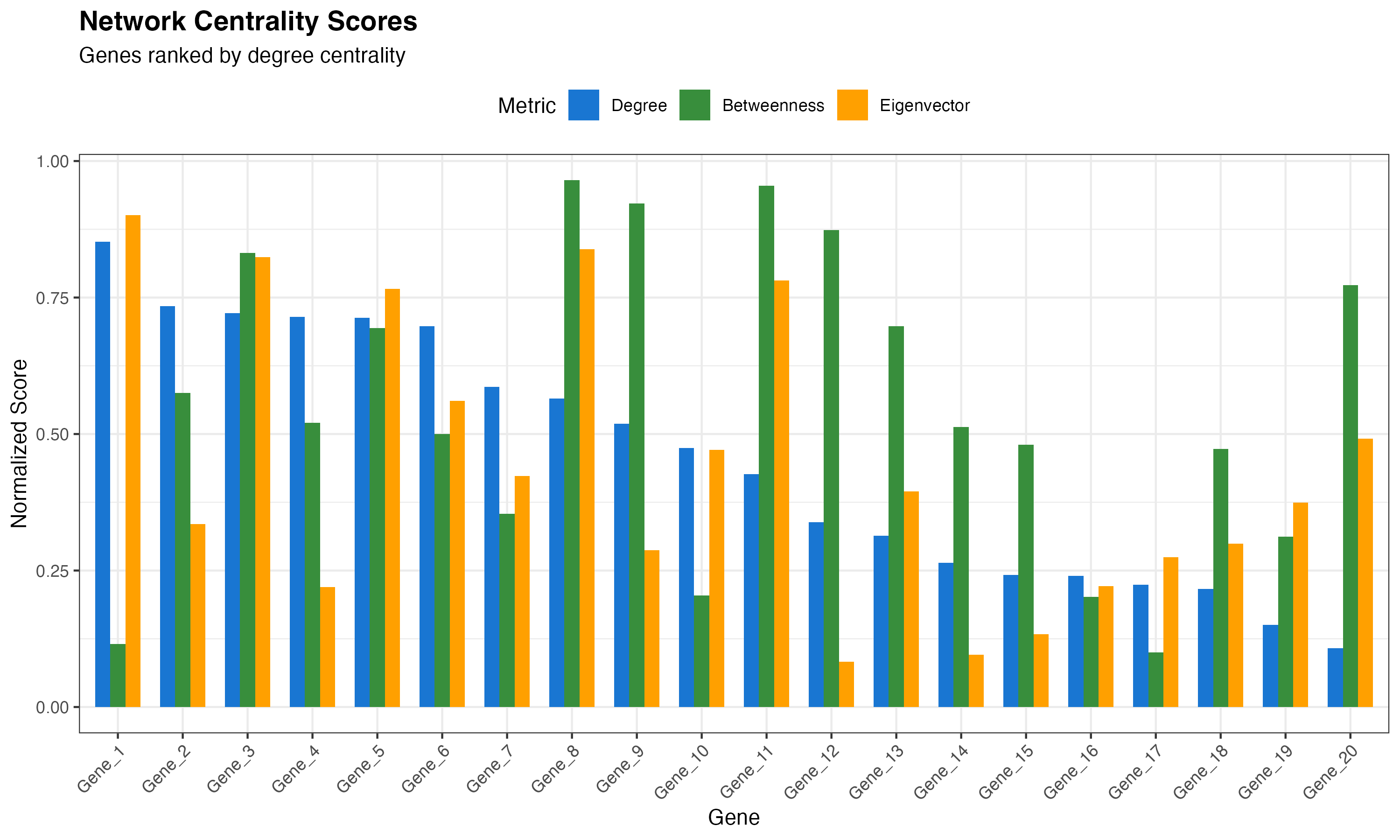
Network Scores Ranking
# Create mock network scores
scores_df <- data.frame(
gene = paste0("Gene_", 1:20),
degree = sort(runif(20, 0, 1), decreasing = TRUE),
betweenness = runif(20, 0, 1),
eigenvector = runif(20, 0, 1)
)
scores_df$gene <- factor(scores_df$gene, levels = scores_df$gene)
# Reshape for plotting
library(reshape2)
scores_long <- melt(scores_df, id.vars = "gene", variable.name = "metric", value.name = "score")
ggplot(scores_long, aes(x = gene, y = score, fill = metric)) +
geom_col(position = "dodge", width = 0.7) +
scale_fill_manual(
values = c("#1976D2", "#388E3C", "#FFA000"),
labels = c("Degree", "Betweenness", "Eigenvector")
) +
labs(
title = "Network Centrality Scores",
subtitle = "Genes ranked by degree centrality",
x = "Gene",
y = "Normalized Score",
fill = "Metric"
) +
theme_bw() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1),
plot.title = element_text(face = "bold", size = 14),
legend.position = "top"
)
Pseudotime Visualizations
Pseudotime on Embedding
# Add pseudotime to demo data
demo_embedding$pseudotime <- with(demo_embedding, {
# Pseudotime based on distance from HSC cluster center
dist_from_origin <- sqrt((UMAP_1 + 3)^2 + UMAP_2^2)
scales::rescale(dist_from_origin, to = c(0, 1))
})
ggplot(demo_embedding, aes(x = UMAP_1, y = UMAP_2, color = pseudotime)) +
geom_point(alpha = 0.7, size = 1.5) +
scale_color_viridis_c(option = "magma", direction = -1) +
labs(
title = "Pseudotime Trajectory",
subtitle = "Color indicates developmental progression",
x = "UMAP 1",
y = "UMAP 2",
color = "Pseudotime"
) +
theme_minimal() +
theme(
panel.grid = element_blank(),
plot.title = element_text(face = "bold", size = 14)
) +
coord_fixed()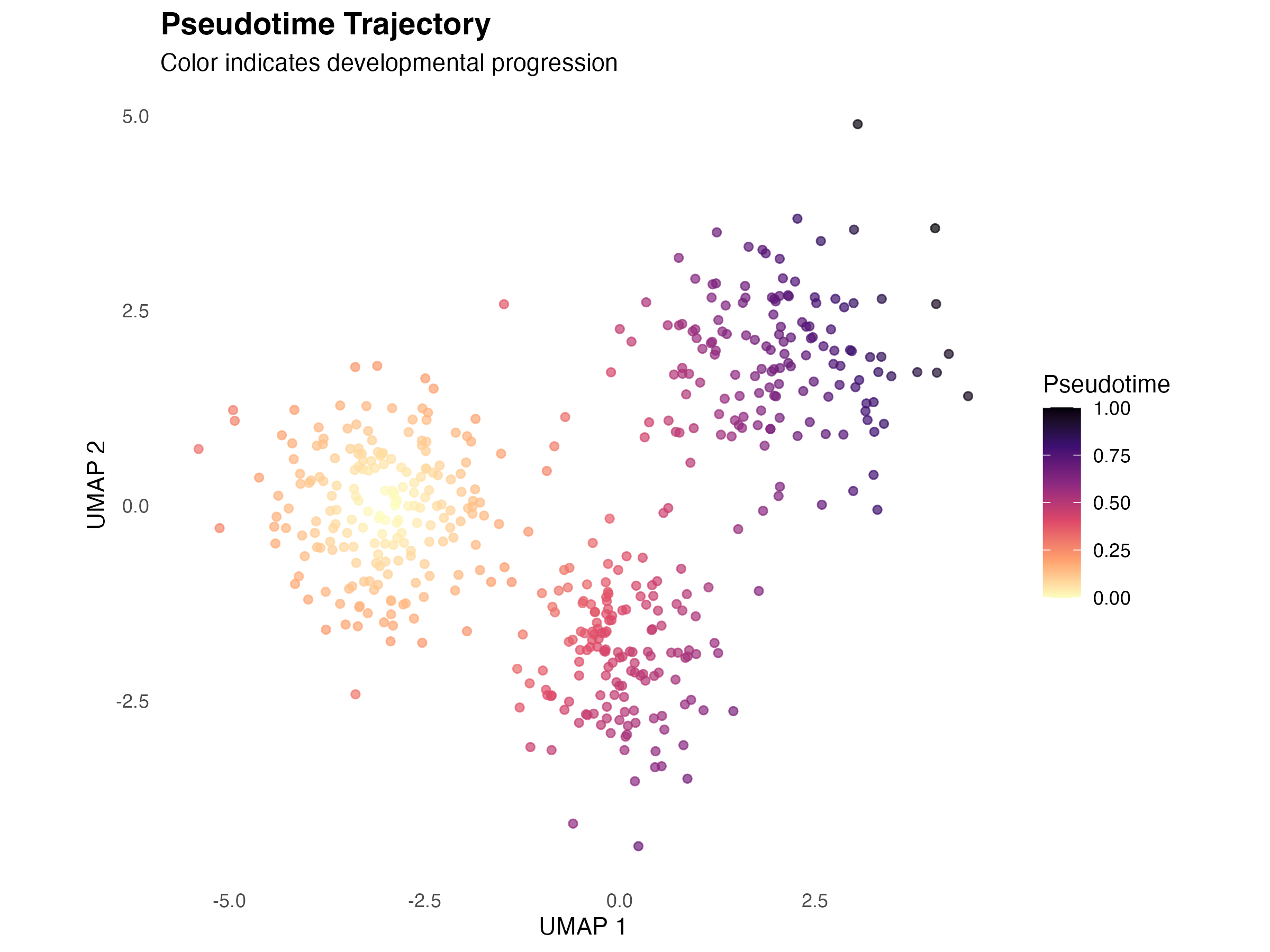
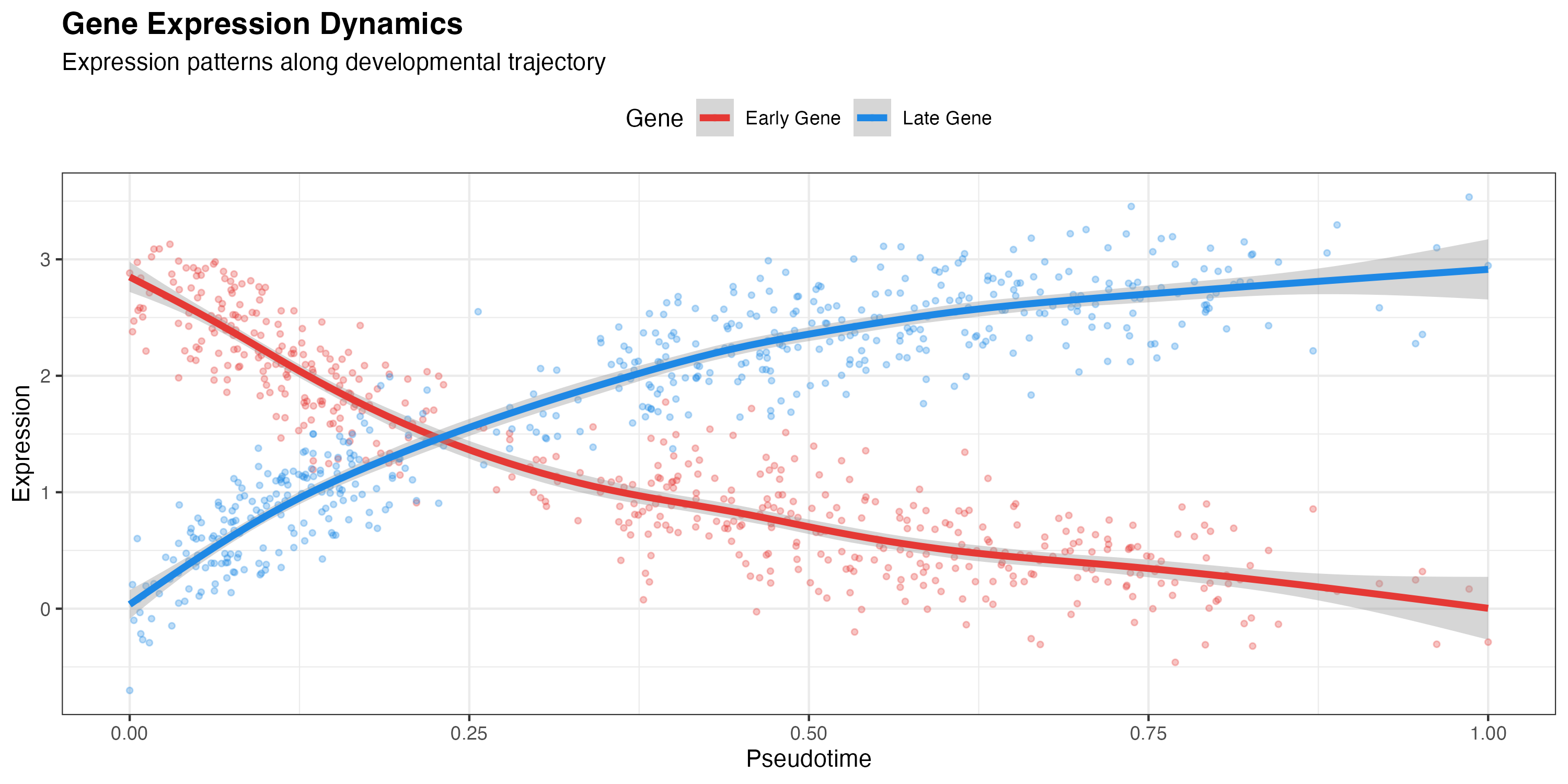
Gene Expression along Pseudotime
# Generate gene expression patterns
demo_embedding$Gene_early <- 3 * exp(-3 * demo_embedding$pseudotime) + rnorm(n_cells, 0, 0.3)
demo_embedding$Gene_late <- 3 * (1 - exp(-3 * demo_embedding$pseudotime)) + rnorm(n_cells, 0, 0.3)
# Reshape for plotting
expr_long <- melt(demo_embedding[, c("pseudotime", "Gene_early", "Gene_late")],
id.vars = "pseudotime",
variable.name = "gene",
value.name = "expression")
ggplot(expr_long, aes(x = pseudotime, y = expression, color = gene)) +
geom_point(alpha = 0.3, size = 1) +
geom_smooth(method = "gam", formula = y ~ s(x, bs = "cs"), size = 1.5) +
scale_color_manual(
values = c("#E53935", "#1E88E5"),
labels = c("Early Gene", "Late Gene")
) +
labs(
title = "Gene Expression Dynamics",
subtitle = "Expression patterns along developmental trajectory",
x = "Pseudotime",
y = "Expression",
color = "Gene"
) +
theme_bw() +
theme(
plot.title = element_text(face = "bold", size = 14),
legend.position = "top"
)
Comparison Visualizations
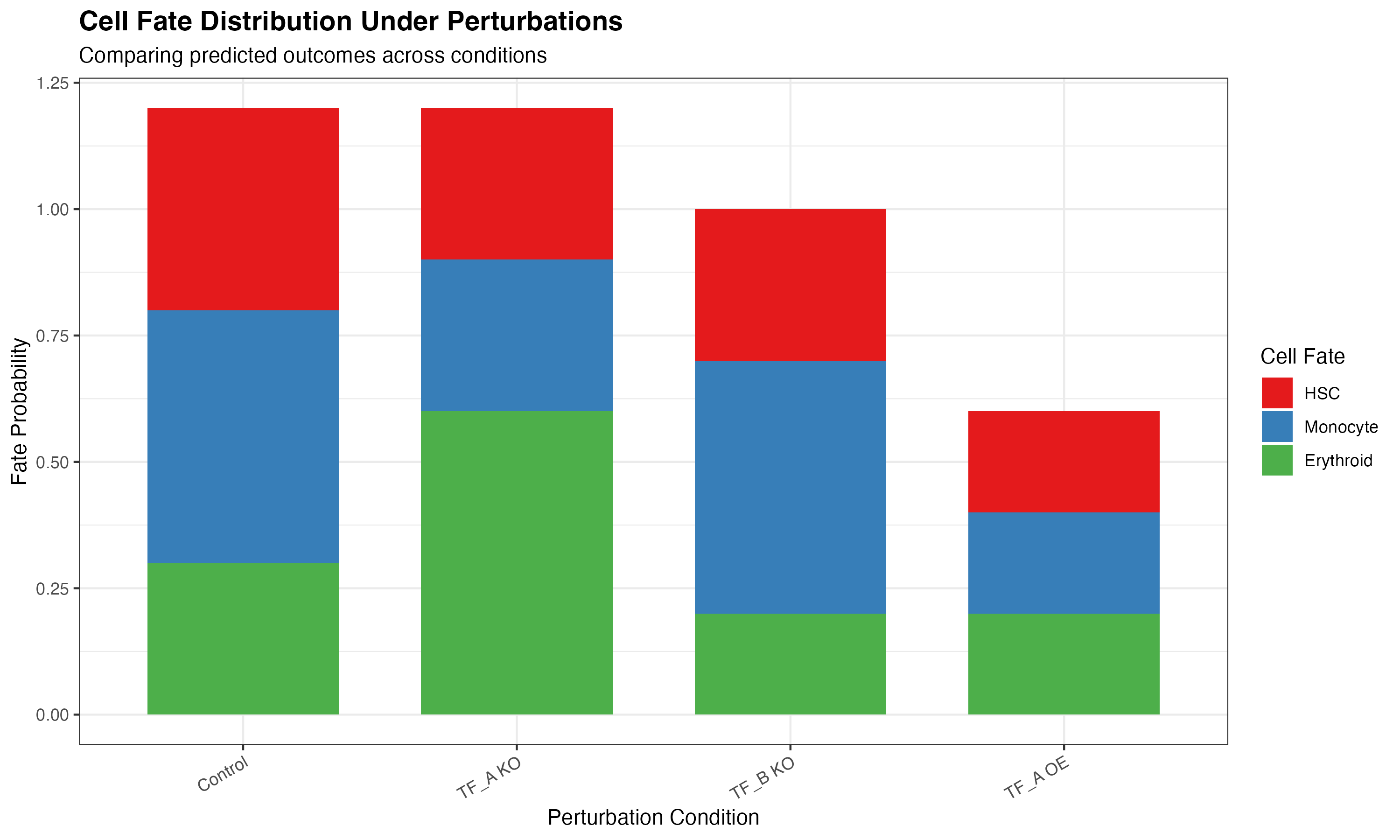
Perturbation Comparison
# Create comparison data
perturbations <- c("Control", "TF_A KO", "TF_B KO", "TF_A OE")
metrics <- c("HSC", "Monocyte", "Erythroid")
comparison_data <- expand.grid(
perturbation = perturbations,
fate = metrics
)
# Simulate fate probabilities
set.seed(42)
comparison_data$probability <- c(
0.4, 0.3, 0.3, # Control
0.2, 0.5, 0.3, # TF_A KO
0.5, 0.2, 0.3, # TF_B KO
0.6, 0.2, 0.2 # TF_A OE
)
ggplot(comparison_data, aes(x = perturbation, y = probability, fill = fate)) +
geom_col(position = "stack", width = 0.7) +
scale_fill_manual(values = c("#E41A1C", "#377EB8", "#4DAF4A")) +
labs(
title = "Cell Fate Distribution Under Perturbations",
subtitle = "Comparing predicted outcomes across conditions",
x = "Perturbation Condition",
y = "Fate Probability",
fill = "Cell Fate"
) +
theme_bw() +
theme(
plot.title = element_text(face = "bold", size = 14),
axis.text.x = element_text(angle = 30, hjust = 1)
)
Customization Guide
Color Palettes
CellOracleR visualizations support custom color schemes:
# Show available color schemes
palettes <- list(
"viridis" = viridis::viridis(10),
"plasma" = viridis::plasma(10),
"magma" = viridis::magma(10),
"inferno" = viridis::inferno(10)
)
par(mfrow = c(1, 4), mar = c(1, 1, 2, 1))
for (name in names(palettes)) {
barplot(rep(1, 10), col = palettes[[name]], border = NA,
main = name, axes = FALSE)
}
Theme Options
# Apply custom themes
library(CellOracleR)
plot_cluster(oracle, cluster_col = "cell_type") +
theme_minimal() +
theme(
legend.position = "bottom",
plot.title = element_text(face = "bold")
)Summary
CellOracleR provides publication-ready visualizations for:
| Function | Purpose |
|---|---|
plot_cluster() |
Cell clusters in embedding |
plot_gene_expression() |
Gene expression overlay |
plot_quiver() |
Vector field of cell movement |
plot_simulation_flow() |
Streamlined flow visualization |
plot_network_graph() |
GRN visualization |
plot_degree_distribution() |
Network topology |
plot_scores_as_rank() |
Gene ranking by network metrics |
plot_pseudotime() |
Developmental trajectory |
plot_simulation_combined() |
Multi-panel summary |
All functions return ggplot2 objects for easy customization and combination.
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] reshape2_1.4.5 patchwork_1.3.2 Matrix_1.7-4 ggplot2_4.0.1
#>
#> loaded via a namespace (and not attached):
#> [1] viridis_0.6.5 sass_0.4.10 generics_0.1.4 stringi_1.8.7
#> [5] lattice_0.22-7 digest_0.6.39 magrittr_2.0.4 evaluate_1.0.5
#> [9] grid_4.4.0 RColorBrewer_1.1-3 fastmap_1.2.0 plyr_1.8.9
#> [13] jsonlite_2.0.0 gridExtra_2.3 mgcv_1.9-3 viridisLite_0.4.2
#> [17] scales_1.4.0 textshaping_1.0.4 jquerylib_0.1.4 cli_3.6.5
#> [21] rlang_1.1.7 splines_4.4.0 withr_3.0.2 cachem_1.1.0
#> [25] yaml_2.3.12 otel_0.2.0 tools_4.4.0 dplyr_1.1.4
#> [29] vctrs_0.7.1 R6_2.6.1 lifecycle_1.0.5 stringr_1.6.0
#> [33] fs_1.6.6 htmlwidgets_1.6.4 ragg_1.5.0 pkgconfig_2.0.3
#> [37] desc_1.4.3 pkgdown_2.1.3 pillar_1.11.1 bslib_0.9.0
#> [41] gtable_0.3.6 glue_1.8.0 Rcpp_1.1.1 systemfonts_1.3.1
#> [45] xfun_0.56 tibble_3.3.1 tidyselect_1.2.1 knitr_1.51
#> [49] dichromat_2.0-0.1 farver_2.1.2 htmltools_0.5.9 nlme_3.1-168
#> [53] rmarkdown_2.30 labeling_0.4.3 compiler_4.4.0 S7_0.2.1