Getting Started with CellOracleR
Complete Workflow for In Silico Gene Perturbation Analysis
Zaoqu Liu
2026-01-25
Source:vignettes/tutorial.Rmd
tutorial.RmdIntroduction
CellOracleR enables in silico gene perturbation analysis of single-cell RNA-seq data. By combining gene regulatory network (GRN) inference with expression data, CellOracleR predicts cell state transitions in response to transcription factor perturbations.
Key Features
- R6 Object-Oriented Design: Clean, intuitive API
- Seurat Integration: Seamless workflow with Seurat V4/V5
- High Performance: C++ acceleration for critical operations
- Publication-Ready Visualizations: ggplot2-based plotting functions
- Comprehensive Network Analysis: Centrality metrics, hub detection, and more
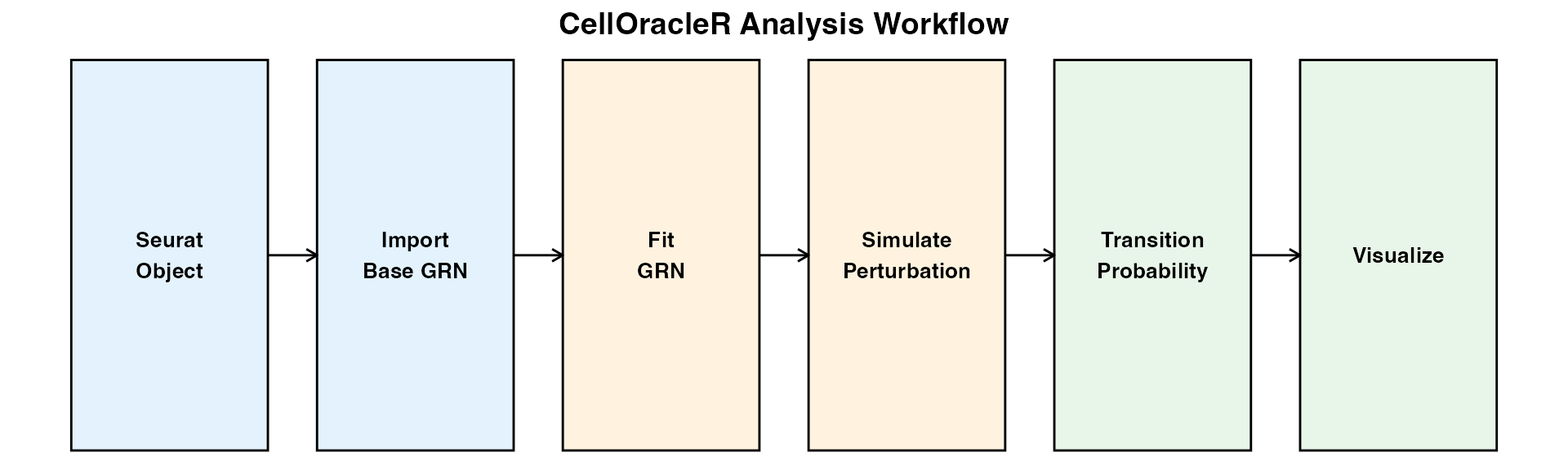
Installation
# Install from R-universe (recommended)
install.packages("CellOracleR", repos = "https://zaoqu-liu.r-universe.dev")
# Or install from GitHub
devtools::install_github("Zaoqu-Liu/CellOracleR")
# For motif analysis, install Bioconductor dependencies
BiocManager::install(c(
"TFBSTools", "motifmatchr",
"BSgenome.Hsapiens.UCSC.hg38", "JASPAR2020"
))Step 1: Prepare Data
CellOracleR works with standard Seurat objects. The data should be normalized (log-transformed) but NOT scaled/centered.
# Load your Seurat object
seurat <- readRDS("path/to/seurat.rds")
# Check data structure
seuratCreating Demo Data
For this tutorial, we’ll create a mock dataset:
set.seed(42)
n_cells <- 1000
n_genes <- 500
# Simulate expression with cluster structure
cluster_means <- list(
c1 = c(rep(3, 50), rep(1, 450)), # Cluster 1: high expression in first 50 genes
c2 = c(rep(1, 50), rep(3, 100), rep(1, 350)), # Cluster 2
c3 = c(rep(1, 150), rep(3, 100), rep(1, 250)) # Cluster 3
)
# Create expression matrix
cells_per_cluster <- 333
expr_list <- list()
for (i in 1:3) {
expr_list[[i]] <- matrix(
rpois(cells_per_cluster * n_genes, lambda = cluster_means[[i]]),
nrow = n_genes
)
}
counts <- do.call(cbind, expr_list)
rownames(counts) <- paste0("Gene", 1:n_genes)
colnames(counts) <- paste0("Cell", 1:ncol(counts))
# Create Seurat object
seurat <- CreateSeuratObject(counts = counts)
seurat <- NormalizeData(seurat)
seurat <- FindVariableFeatures(seurat)
seurat <- ScaleData(seurat)
seurat <- RunPCA(seurat)
seurat <- FindNeighbors(seurat, dims = 1:30)
seurat <- FindClusters(seurat, resolution = 0.5)
seurat <- RunUMAP(seurat, dims = 1:30)Step 2: Create Oracle Object
# Create Oracle from Seurat
oracle <- create_oracle(
seurat = seurat,
cluster_column = "seurat_clusters",
embedding_name = "umap",
verbose = TRUE
)
# Print summary
oracleOutput:
CellOracleR Oracle Object
Cells: 1000
Genes: 500
Clusters: 3 (0, 1, 2)
Embedding: UMAP (2D)
GRN fitted: FALSE
Simulation done: FALSEStep 3: Import Base GRN
The base GRN defines which TFs can potentially regulate which genes. Options:
Option A: From Motif Analysis (Recommended)
# Requires scATAC-seq peak data
base_grn <- create_base_grn(
peaks_df = peaks_df, # data.frame with chr, start, end, gene_short_name
ref_genome = "hg38",
upstream = 2000,
downstream = 2000,
fpr = 0.02,
n_cores = 4
)Option B: From Pre-built Database
# Load from CSV file
base_grn <- load_base_grn("tf_target_database.csv")Option C: Custom Dictionary
# Define regulatory relationships manually
# List format: target_gene -> c(regulator1, regulator2, ...)
TFdict <- list(
Gene1 = c("Gene10", "Gene20", "Gene30"),
Gene2 = c("Gene10", "Gene15", "Gene25"),
Gene3 = c("Gene20", "Gene30", "Gene40")
)
# For demo: create simple TF dictionary
TFdict <- lapply(paste0("Gene", 1:100), function(g) {
sample(paste0("Gene", 101:200), size = sample(3:8, 1))
})
names(TFdict) <- paste0("Gene", 1:100)Step 4: Preprocessing
# Use Seurat's PCA or compute new
oracle$perform_PCA(
n_components = 50,
use_seurat_pca = TRUE # Use existing PCA from Seurat
)
# KNN imputation for noise reduction (optional but recommended)
oracle$knn_imputation(
k = 30, # Number of neighbors (NULL for auto)
n_pca_dims = 50 # PCA dimensions for neighbor search
)Step 5: Fit Cluster-Specific GRN
This is the core step - fitting regulatory coefficients using Ridge regression:
oracle$fit_grn_for_simulation(
GRN_unit = "cluster", # Fit separate GRN per cluster
alpha = 10, # Ridge regularization (1-100)
bagging_number = 200, # Bootstrap iterations
verbose = TRUE
)Key Parameters:
| Parameter | Description | Recommended |
|---|---|---|
GRN_unit |
“cluster” or “whole” | “cluster” |
alpha |
Regularization strength | 10 |
bagging_number |
Bootstrap iterations | 200 |
Step 6: Simulate Perturbation
Now we can simulate the effects of gene perturbations:
# Define perturbation: Gene10 knockout (expression = 0)
perturb_condition <- list(Gene10 = 0)
# Or use helper function
perturb_condition <- create_perturb_condition(
genes = "Gene10",
expression = 0
)
# Run simulation
oracle$simulate_shift(
perturb_condition = perturb_condition,
n_propagation = 3 # Signal propagation steps (1-5)
)
# View summary
simulation_summary(oracle)Perturbation Types:
# Knockout (KO): Set expression to 0
perturb_ko <- list(Gene10 = 0)
# Overexpression (OE): Set to maximum observed value
max_expr <- max(oracle$adata$layers$imputed_count["Gene10", ])
perturb_oe <- list(Gene10 = max_expr)
# Multiple perturbations
perturb_multi <- list(
Gene10 = 0, # KO Gene10
Gene20 = max_expr # OE Gene20
)Step 7: Calculate Transition Probabilities
# Compute transition probabilities
oracle$estimate_transition_prob(
k = 200, # KNN neighbors
sampled_fraction = 1, # Cell sampling fraction
calculate_randomized = TRUE
)
# Calculate embedding shifts
oracle$calculate_embedding_shift()
# Calculate grid arrows for visualization
oracle$calculate_grid_arrows(
n_grid = 40,
n_neighbors = 100,
smooth = 0.5
)Step 8: Visualize Results
Simulation Flow Field
The main visualization showing predicted cell state transitions:
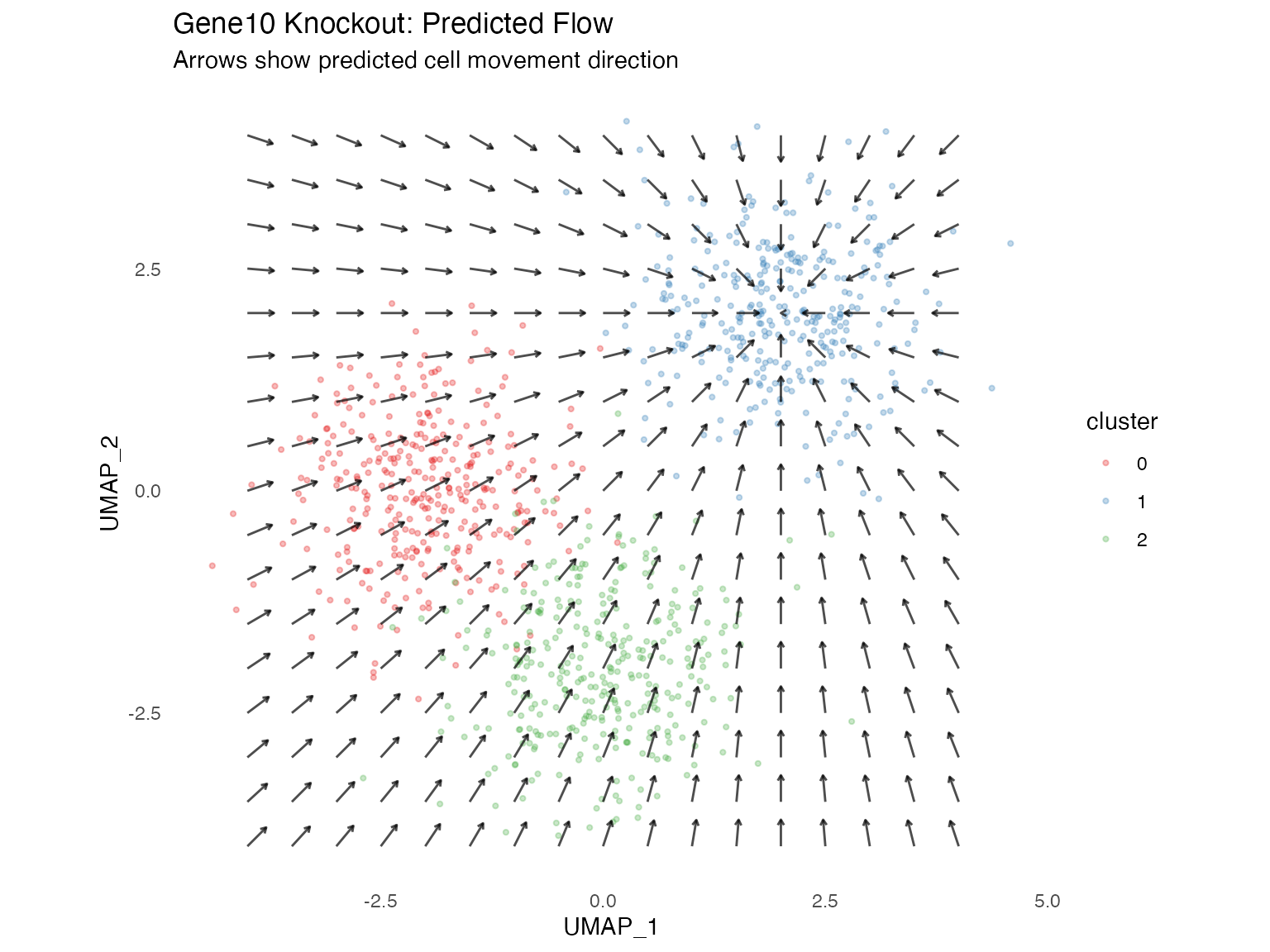
# Main result visualization
plot_simulation_flow(
oracle,
scale = 30,
min_mass = 0.01,
arrow_color = "black",
title = "Gene10 KO: Predicted Cell Transitions"
)Gene Expression Changes
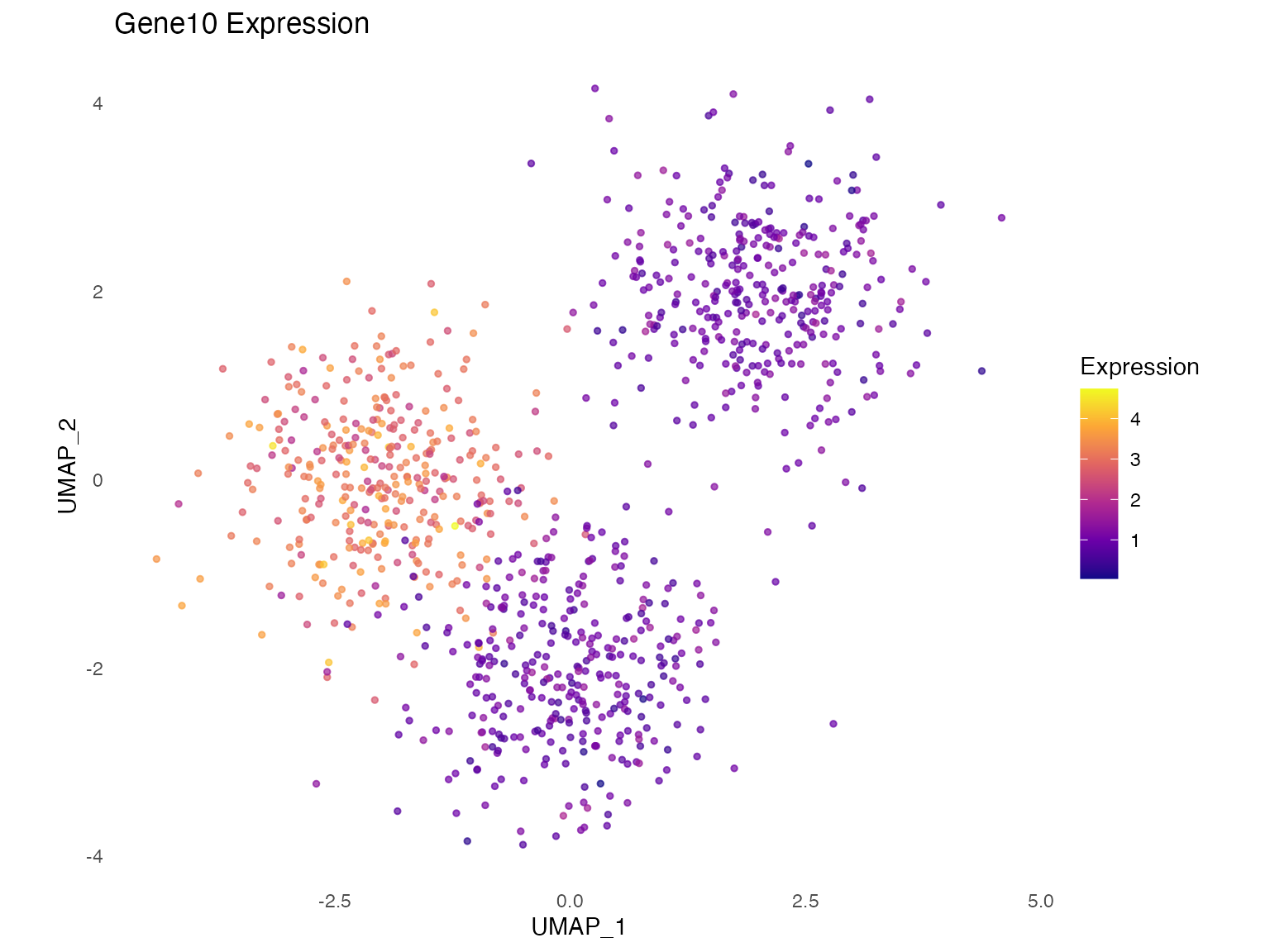
# Before perturbation
plot_gene_expression(oracle, "Gene10", title = "Gene10 Expression")
# Simulated change
plot_gene_expression(oracle, "Gene10", layer = "delta_X",
title = "Gene10 Change (Delta)")Step 9: Network Analysis
For detailed network characterization:
# Get Links object
links <- oracle$get_links(
cluster_name_for_GRN_unit = "0", # Cluster name
alpha = 10,
bagging_number = 200
)
# Filter by significance
links$filter(
threshold_p = 0.001,
threshold_coef = 0.1
)
# Calculate network metrics
links$get_network_score()
# View network summary
print(links)Top Regulators
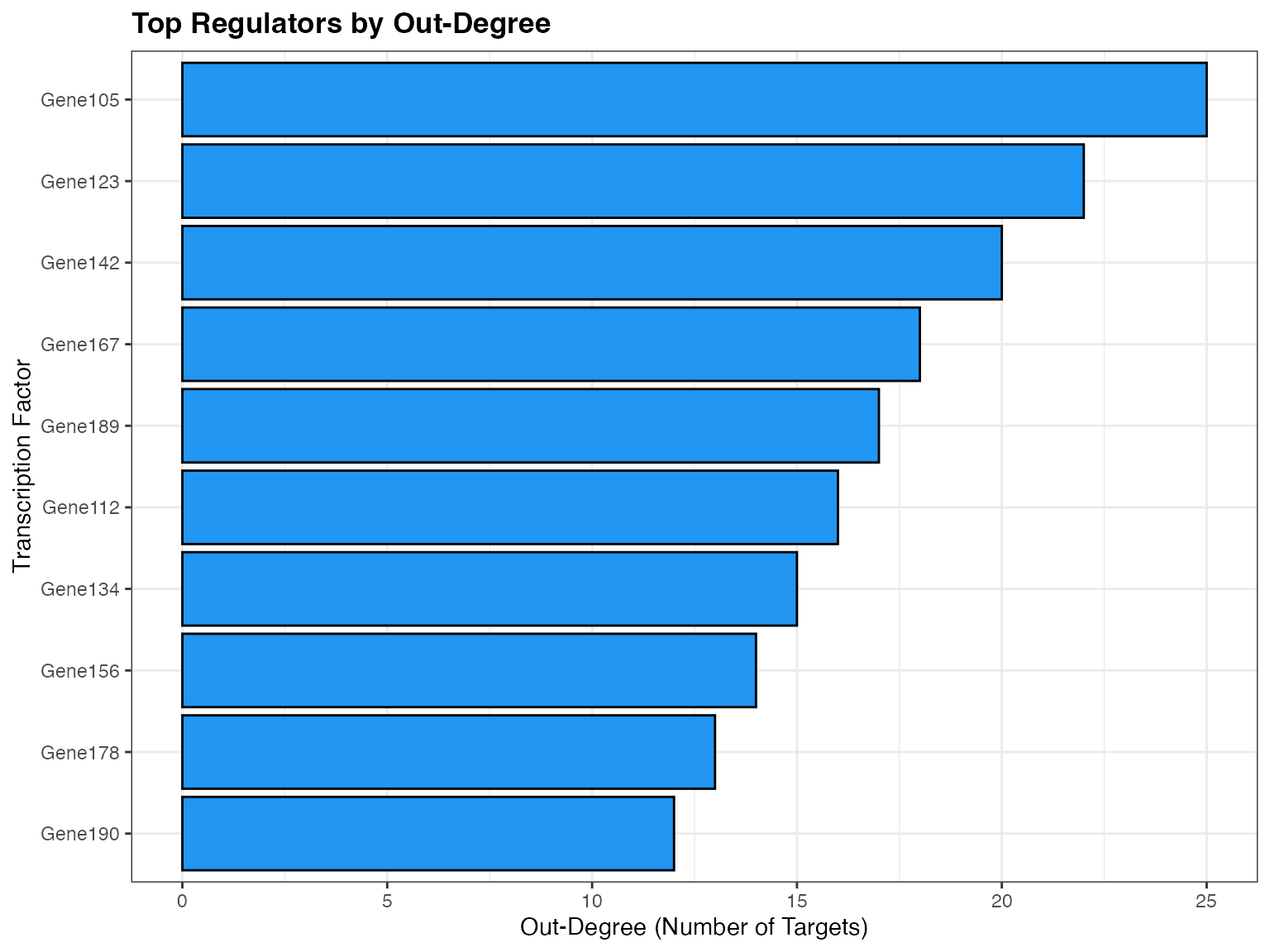
# Rank TFs by network metrics
plot_scores_as_rank(links, metric = "degree_out", top_n = 20)Saving and Loading
# Save Oracle object (uses qs for fast serialization)
save_oracle(oracle, "my_analysis.qs")
# Load Oracle object
oracle <- load_oracle("my_analysis.qs")Advanced: Comparing Multiple Perturbations
# List of genes to perturb
genes_to_test <- c("Gene10", "Gene20", "Gene30")
# Run systematic analysis
plots <- list()
for (gene in genes_to_test) {
# Create copy to avoid overwriting
oracle_test <- oracle$clone(deep = TRUE)
# Simulate knockout
oracle_test$simulate_shift(
perturb_condition = list(gene = 0),
n_propagation = 3
)
# Calculate transitions
oracle_test$estimate_transition_prob()
oracle_test$calculate_embedding_shift()
oracle_test$calculate_grid_arrows()
# Store plot
plots[[gene]] <- plot_simulation_flow(oracle_test, title = paste(gene, "KO"))
}
# Combine plots
library(patchwork)
wrap_plots(plots, ncol = 2)Summary
You’ve learned how to:
- ✅ Create an Oracle object from Seurat data
- ✅ Import regulatory network priors
- ✅ Fit cluster-specific GRNs
- ✅ Simulate gene perturbations
- ✅ Calculate transition probabilities
- ✅ Visualize predicted cell fate changes
- ✅ Perform network analysis
Next Steps
- Read the Algorithm Principles vignette for mathematical details
- Explore the Visualization Gallery for more plot types
- See the Case Study for a complete biological example
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggplot2_4.0.1
#>
#> loaded via a namespace (and not attached):
#> [1] gtable_0.3.6 jsonlite_2.0.0 dplyr_1.1.4 compiler_4.4.0
#> [5] tidyselect_1.2.1 dichromat_2.0-0.1 jquerylib_0.1.4 systemfonts_1.3.1
#> [9] scales_1.4.0 textshaping_1.0.4 yaml_2.3.12 fastmap_1.2.0
#> [13] R6_2.6.1 labeling_0.4.3 generics_0.1.4 knitr_1.51
#> [17] htmlwidgets_1.6.4 tibble_3.3.1 desc_1.4.3 bslib_0.9.0
#> [21] pillar_1.11.1 RColorBrewer_1.1-3 rlang_1.1.7 cachem_1.1.0
#> [25] xfun_0.56 fs_1.6.6 sass_0.4.10 S7_0.2.1
#> [29] otel_0.2.0 viridisLite_0.4.2 cli_3.6.5 pkgdown_2.1.3
#> [33] withr_3.0.2 magrittr_2.0.4 digest_0.6.39 grid_4.4.0
#> [37] lifecycle_1.0.5 vctrs_0.7.1 evaluate_1.0.5 glue_1.8.0
#> [41] farver_2.1.2 ragg_1.5.0 rmarkdown_2.30 tools_4.4.0
#> [45] pkgconfig_2.0.3 htmltools_0.5.9