Building Custom References
Zaoqu Liu
2026-01-26
Source:vignettes/custom-reference.Rmd
custom-reference.RmdIntroduction
While CellProgramMapper provides pre-built references for common cell types, you may want to create custom references for:
- Novel cell types or tissues
- Specific biological contexts
- Combining multiple datasets
- Custom gene expression programs
This vignette covers the process of building and using custom references.
Reference Format
A reference consists of a spectra matrix where:
- Rows: Gene expression programs (GEPs)
- Columns: Genes
- Values: Non-negative weights representing gene contributions
Method 1: From cNMF Results
Running cNMF
First, run cNMF (consensus Non-negative Matrix Factorization) on your reference data using Python:
# In Python
from cnmf import cNMF
import scanpy as sc
# Load data
adata = sc.read_h5ad("reference_data.h5ad")
# Initialize cNMF
cnmf_obj = cNMF(output_dir='./cnmf_output', name='my_reference')
# Prepare data
cnmf_obj.prepare(counts_fn=adata, components=np.arange(5, 25),
n_iter=200, seed=42)
# Run factorization
cnmf_obj.factorize(worker_i=0, total_workers=1)
# Compute consensus
cnmf_obj.consensus(k=15, density_threshold=0.1)Loading cNMF Results
library(CellProgramMapper)
# Load from cNMF output directory
spectra <- load_cnmf_spectra(
cnmf_dir = "./cnmf_output",
cnmf_name = "my_reference",
k = 15 # Number of programs
)
# Use as reference
result <- CellProgramMapper(
query = seurat_obj,
reference = spectra
)Method 2: Building Consensus Reference
When you have cNMF results from multiple datasets, use
BuildConsensusReference to create a unified reference:
# Initialize builder
builder <- BuildConsensusReference(
output_dir = "./consensus_output",
name = "my_consensus_reference"
)
# Add cNMF results from multiple datasets
builder$add_cnmf_result(
cnmf_dir = "./dataset1/cnmf_output",
cnmf_name = "dataset1",
k = 15
)
builder$add_cnmf_result(
cnmf_dir = "./dataset2/cnmf_output",
cnmf_name = "dataset2",
k = 12
)
# Compute correlations between all GEPs
builder$compute_gep_correlations()
# Cluster GEPs into consensus programs
builder$cluster_geps(
correlation_threshold = 0.5,
min_cluster_size = 2
)
# Get final consensus spectra
consensus_spectra <- builder$get_consensus_spectra()Method 3: Manual Construction
From Known Gene Signatures
library(CellProgramMapper)
#> CellProgramMapper v1.0.0
#> Map single cells to reference gene expression programs
#> GitHub: https://github.com/Zaoqu-Liu/CellProgramMapper
# Define gene signatures
signatures <- list(
Exhaustion = c("PDCD1", "LAG3", "HAVCR2", "TIGIT", "CTLA4"),
Cytotoxicity = c("GZMA", "GZMB", "PRF1", "GNLY", "NKG7"),
Proliferation = c("MKI67", "TOP2A", "PCNA", "CDK1", "CCNB1"),
Memory = c("IL7R", "TCF7", "LEF1", "CCR7", "SELL")
)
# Create binary spectra
all_genes <- unique(unlist(signatures))
spectra <- matrix(0, nrow = length(signatures), ncol = length(all_genes))
rownames(spectra) <- names(signatures)
colnames(spectra) <- all_genes
for (i in seq_along(signatures)) {
spectra[i, signatures[[i]]] <- 1
}
# View spectra
print(spectra)
#> PDCD1 LAG3 HAVCR2 TIGIT CTLA4 GZMA GZMB PRF1 GNLY NKG7 MKI67
#> Exhaustion 1 1 1 1 1 0 0 0 0 0 0
#> Cytotoxicity 0 0 0 0 0 1 1 1 1 1 0
#> Proliferation 0 0 0 0 0 0 0 0 0 0 1
#> Memory 0 0 0 0 0 0 0 0 0 0 0
#> TOP2A PCNA CDK1 CCNB1 IL7R TCF7 LEF1 CCR7 SELL
#> Exhaustion 0 0 0 0 0 0 0 0 0
#> Cytotoxicity 0 0 0 0 0 0 0 0 0
#> Proliferation 1 1 1 1 0 0 0 0 0
#> Memory 0 0 0 0 1 1 1 1 1From Expression Data
# Simulate expression data for demonstration
set.seed(42)
n_cells <- 200
n_genes <- 50
# Create expression matrix
expression <- matrix(rpois(n_cells * n_genes, lambda = 5),
nrow = n_cells, ncol = n_genes)
colnames(expression) <- paste0("Gene", 1:n_genes)
rownames(expression) <- paste0("Cell", 1:n_cells)
# Define cell type labels
cell_types <- rep(c("TypeA", "TypeB", "TypeC", "TypeD"), each = 50)
# Compute mean expression per cell type
spectra <- do.call(rbind, lapply(unique(cell_types), function(ct) {
cells <- which(cell_types == ct)
colMeans(expression[cells, ])
}))
rownames(spectra) <- unique(cell_types)
# Ensure non-negativity
spectra[spectra < 0] <- 0
print(dim(spectra))
#> [1] 4 50Validating Custom References
Quality Checks
validate_reference <- function(spectra) {
checks <- list()
# Check 1: Non-negativity
checks$non_negative <- all(spectra >= 0)
# Check 2: No all-zero programs
checks$no_zero_programs <- all(rowSums(spectra) > 0)
# Check 3: No all-zero genes
checks$no_zero_genes <- all(colSums(spectra) > 0)
# Check 4: Reasonable number of programs
checks$reasonable_k <- nrow(spectra) >= 2 && nrow(spectra) <= 100
# Check 5: Sufficient genes
checks$sufficient_genes <- ncol(spectra) >= 100
# Report
cat("Reference validation:\n")
for (check_name in names(checks)) {
status <- if (checks[[check_name]]) "PASS" else "FAIL"
cat(sprintf(" %s: %s\n", check_name, status))
}
return(all(unlist(checks)))
}
# Note: Our demo spectra has fewer genes than recommended
# validate_reference(spectra)Visualize Reference
# Heatmap of spectra
if (!requireNamespace("pheatmap", quietly = TRUE)) {
install.packages("pheatmap")
}
# For demonstration, use the signature-based spectra
library(pheatmap)
#> Warning: package 'pheatmap' was built under R version 4.4.1
pheatmap(
spectra,
cluster_rows = TRUE,
cluster_cols = TRUE,
show_colnames = TRUE,
main = "Custom Reference Spectra",
color = colorRampPalette(c("white", "#08306b"))(100)
)
Visualization of reference spectra
Saving References
As TSV File
# Save spectra matrix
write.table(spectra, file = "my_reference.tsv",
sep = "\t", quote = FALSE, row.names = TRUE)With Metadata
# Save with additional information
reference_data <- list(
spectra = spectra,
metadata = list(
name = "My Custom Reference",
version = "1.0",
description = "Reference for XYZ cell types",
species = "Homo sapiens",
source_datasets = c("Dataset1", "Dataset2"),
date_created = Sys.Date()
)
)
saveRDS(reference_data, "my_reference.rds")Using Custom References
# Method 1: From file path
result <- CellProgramMapper(
query = seurat_obj,
reference = "path/to/my_reference.tsv"
)
# Method 2: From matrix object
result <- CellProgramMapper(
query = seurat_obj,
reference = spectra # Your spectra matrix
)
# Method 3: From RDS file
ref_data <- readRDS("my_reference.rds")
result <- CellProgramMapper(
query = seurat_obj,
reference = ref_data$spectra
)Best Practices
Reference Construction
- Use diverse data: Include cells from multiple donors/conditions
- Quality control: Remove low-quality cells before NMF
- Appropriate k: Choose number of programs based on biological complexity
- Gene selection: Use highly variable genes (1000-5000)
Gene Overlap
# Simulate query genes
query_genes <- c(paste0("Gene", 1:40), paste0("OtherGene", 1:20))
ref_genes <- colnames(spectra)
# Compute overlap
overlap <- intersect(query_genes, ref_genes)
query_only <- setdiff(query_genes, ref_genes)
ref_only <- setdiff(ref_genes, query_genes)
cat(sprintf("Query genes: %d\n", length(query_genes)))
#> Query genes: 60
cat(sprintf("Reference genes: %d\n", length(ref_genes)))
#> Reference genes: 50
cat(sprintf("Overlap: %d (%.1f%%)\n", length(overlap),
100 * length(overlap) / length(ref_genes)))
#> Overlap: 40 (80.0%)
# Visualize
par(mar = c(2, 2, 2, 2))
venn_counts <- c(
"Query only" = length(query_only),
"Reference only" = length(ref_only),
"Overlap" = length(overlap)
)
barplot(venn_counts, col = c("#e41a1c", "#377eb8", "#4daf4a"),
main = "Gene Overlap", ylab = "Number of genes")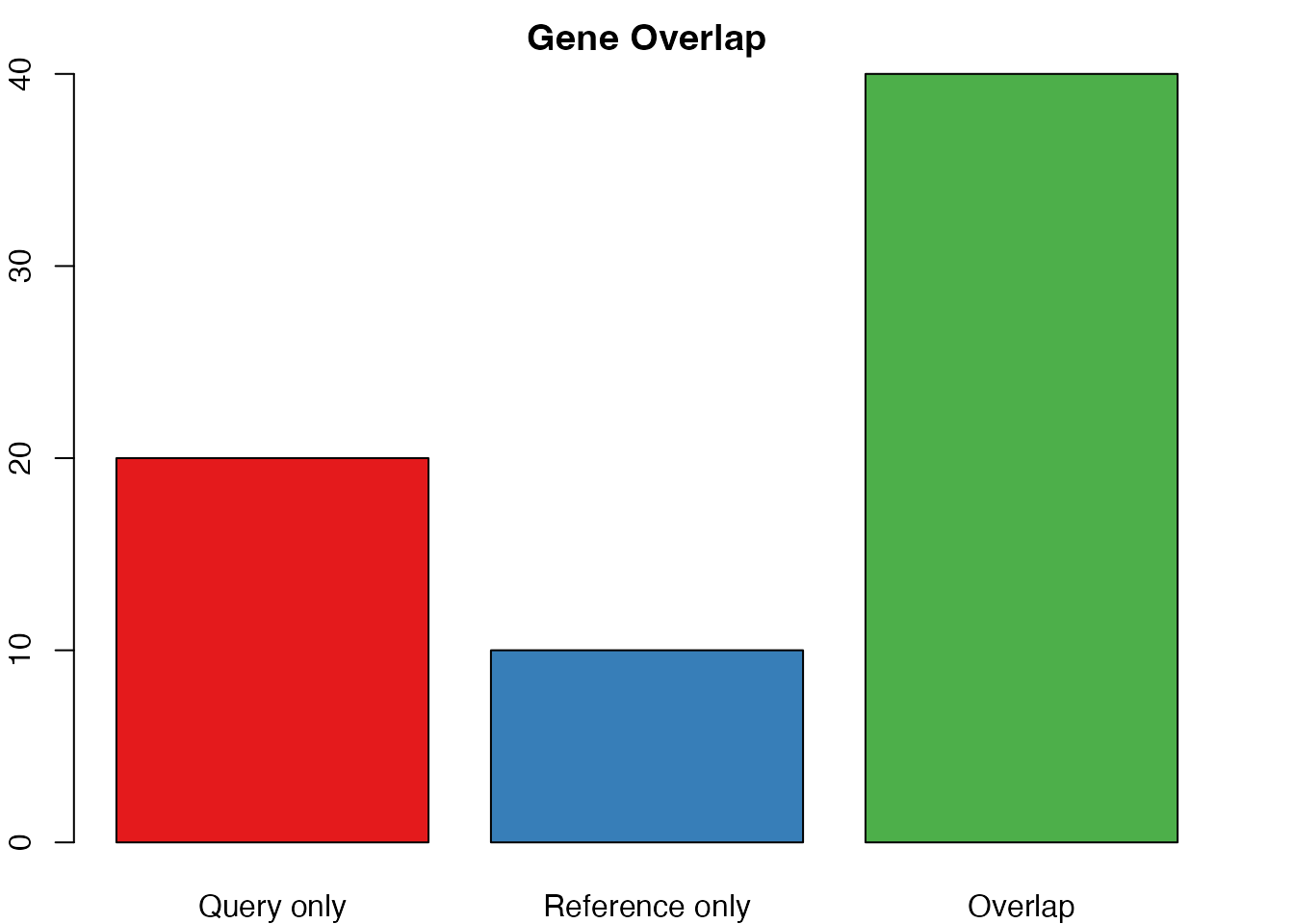
Checking gene overlap between query and reference
Troubleshooting
Low Gene Overlap
# Check overlap
ref_genes <- colnames(spectra)
query_genes <- rownames(GetAssayData(seurat_obj))
overlap <- intersect(query_genes, ref_genes)
if (length(overlap) / length(ref_genes) < 0.5) {
warning("Low gene overlap - check gene naming conventions")
# Try converting gene symbols
# e.g., from ENSEMBL to symbol, or uppercase/lowercase
}All-Zero Usage
If all usage values are zero:
- Check gene overlap (see above)
- Verify data is non-negative
- Check for extreme sparsity
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] pheatmap_1.0.13 CellProgramMapper_1.0.0
#>
#> loaded via a namespace (and not attached):
#> [1] Matrix_1.7-4 gtable_0.3.6 future.apply_1.20.1
#> [4] jsonlite_2.0.0 compiler_4.4.0 Rcpp_1.1.1
#> [7] dichromat_2.0-0.1 parallel_4.4.0 jquerylib_0.1.4
#> [10] scales_1.4.0 globals_0.18.0 systemfonts_1.3.1
#> [13] textshaping_1.0.4 yaml_2.3.12 fastmap_1.2.0
#> [16] lattice_0.22-7 R6_2.6.1 curl_7.0.0
#> [19] knitr_1.51 htmlwidgets_1.6.4 future_1.69.0
#> [22] desc_1.4.3 bslib_0.9.0 RColorBrewer_1.1-3
#> [25] rlang_1.1.7 cachem_1.1.0 xfun_0.56
#> [28] fs_1.6.6 sass_0.4.10 otel_0.2.0
#> [31] cli_3.6.5 pkgdown_2.2.0 digest_0.6.39
#> [34] grid_4.4.0 rappdirs_0.3.4 lifecycle_1.0.5
#> [37] glue_1.8.0 evaluate_1.0.5 data.table_1.18.0
#> [40] farver_2.1.2 listenv_0.10.0 codetools_0.2-20
#> [43] ragg_1.5.0 parallelly_1.46.1 rmarkdown_2.30
#> [46] tools_4.4.0 htmltools_0.5.9