Introduction
CellProgramMapper maps single-cell RNA sequencing data to reference gene expression programs (GEPs) using non-negative matrix factorization. This guide demonstrates the essential workflow in 5 minutes.
Installation
# From R-universe (recommended)
install.packages("CellProgramMapper",
repos = "https://zaoqu-liu.r-universe.dev")
# Or from GitHub
devtools::install_github("Zaoqu-Liu/CellProgramMapper")Quick Example
library(CellProgramMapper)
# Map a Seurat object to T-cell reference
result <- CellProgramMapper(
query = seurat_obj,
reference = "TCAT.V1"
)
# View results
print(result)
# Get usage matrix
usage <- get_usage(result, normalized = TRUE)
# Add to Seurat object
seurat_obj <- add_results_to_seurat(seurat_obj, result)Available References
library(CellProgramMapper)
#> CellProgramMapper v1.0.0
#> Map single cells to reference gene expression programs
#> GitHub: https://github.com/Zaoqu-Liu/CellProgramMapper
refs <- available_references()
print(refs[, c("Name", "Cell_Type", "Species")])
#> Name Cell_Type Species
#> 1 TCAT.V1 T-cells Homo sapiens
#> 2 MYELOID.GLIOMA.V1 Myeloid Homo sapiens
#> 3 BONEMARROW.CD34POS.HSPC.V1 HSC Homo sapiensInput Formats
CellProgramMapper accepts multiple input types:
# 1. Seurat object (V4 or V5)
result <- CellProgramMapper(query = seurat_obj, reference = "TCAT.V1")
# 2. Matrix (cells × genes)
result <- CellProgramMapper(query = counts_matrix, reference = "TCAT.V1")
# 3. File path (h5ad, mtx)
result <- CellProgramMapper(query = "data.h5ad", reference = "TCAT.V1")Working with Results
Access Scores
# Get computed scores
scores <- get_scores(result)Save Results
save_results(result, output_dir = "./output", prefix = "my_analysis")Demonstration with Simulated Data
set.seed(42)
# Simulate reference (5 programs × 100 genes)
H <- matrix(runif(5 * 100, 0, 1), nrow = 5)
colnames(H) <- paste0("Gene", 1:100)
rownames(H) <- paste0("GEP", 1:5)
# Simulate query (50 cells × 100 genes)
W_true <- matrix(runif(50 * 5, 0, 1), nrow = 50)
X <- W_true %*% H + matrix(rnorm(50 * 100, 0, 0.1), nrow = 50)
X[X < 0] <- 0
colnames(X) <- paste0("Gene", 1:100)
rownames(X) <- paste0("Cell", 1:50)
# Run CellProgramMapper
result <- CellProgramMapper(
query = X,
reference = H,
verbose = FALSE
)
#> Warning: Query data does not appear to be integer counts. For best results,
#> provide raw UMI/read counts.
# Visualize
usage <- get_usage(result, normalized = TRUE)
usage_mat <- as.matrix(usage)
par(mfrow = c(1, 2), mar = c(4, 4, 2, 1))
# Heatmap
image(t(usage_mat), col = colorRampPalette(c("white", "#08306b"))(100),
xlab = "Programs", ylab = "Cells", main = "Usage Matrix",
axes = FALSE)
axis(1, at = seq(0, 1, length.out = 5), labels = colnames(usage_mat))
# Bar plot for first cell
barplot(as.numeric(usage[1, ]), col = "#1976d2",
names.arg = colnames(usage),
main = paste("Cell1 Usage"),
xlab = "GEP", ylab = "Usage")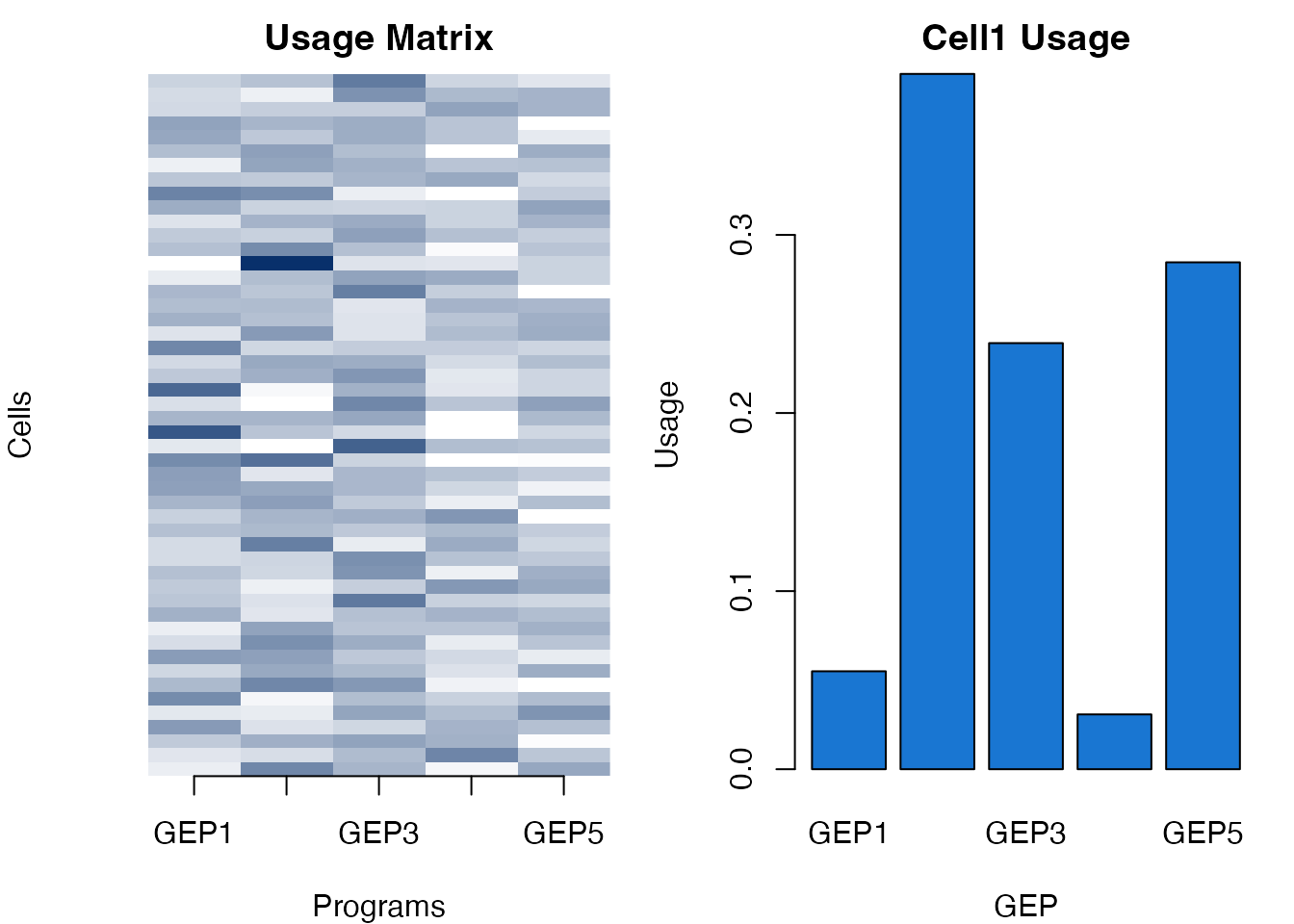
Simulated GEP usage visualization
Performance Tips
# For large datasets, use parallel processing
result <- CellProgramMapper(
query = seurat_obj,
reference = "TCAT.V1",
n_workers = 4
)
# Data is automatically batched for memory efficiencyNext Steps
- Mathematical Framework - Understand the algorithm
- NNLS Solver Details - Implementation details
- Visualization Guide - Create publication figures
- Custom References - Build your own references
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] CellProgramMapper_1.0.0
#>
#> loaded via a namespace (and not attached):
#> [1] cli_3.6.5 knitr_1.51 rlang_1.1.7
#> [4] xfun_0.56 otel_0.2.0 textshaping_1.0.4
#> [7] data.table_1.18.0 jsonlite_2.0.0 future.apply_1.20.1
#> [10] listenv_0.10.0 htmltools_0.5.9 ragg_1.5.0
#> [13] sass_0.4.10 rappdirs_0.3.4 rmarkdown_2.30
#> [16] grid_4.4.0 evaluate_1.0.5 jquerylib_0.1.4
#> [19] fastmap_1.2.0 yaml_2.3.12 lifecycle_1.0.5
#> [22] compiler_4.4.0 codetools_0.2-20 fs_1.6.6
#> [25] Rcpp_1.1.1 htmlwidgets_1.6.4 future_1.69.0
#> [28] systemfonts_1.3.1 lattice_0.22-7 digest_0.6.39
#> [31] R6_2.6.1 parallelly_1.46.1 parallel_4.4.0
#> [34] curl_7.0.0 bslib_0.9.0 Matrix_1.7-4
#> [37] tools_4.4.0 globals_0.18.0 pkgdown_2.2.0
#> [40] cachem_1.1.0 desc_1.4.3