Introduction
This vignette covers advanced usage scenarios for scPharm, including:
- Custom threshold calibration
- Multi-drug analysis strategies
- Combination therapy optimization
- Integration with external tools
- Performance tuning
1. Threshold Calibration with Normal Tissue
Using scPharmGenNullDist
For accurate cell classification, calibrating thresholds using normal tissue samples is recommended:
library(scPharm)
# Load normal tissue reference
normal_seurat <- readRDS("normal_tissue.rds")
# Generate null distribution
null_dist <- scPharmGenNullDist(
normal_seurat,
cancer = "LUAD",
drug = "Erlotinib",
nmcs = 50,
nfeatures = 200
)
# Extract calibrated thresholds
threshold_s <- null_dist$threshold.s
threshold_r <- null_dist$threshold.r
cat("Sensitive threshold:", threshold_s, "\n")
cat("Resistant threshold:", threshold_r, "\n")Apply Calibrated Thresholds
# Load tumor sample
tumor_seurat <- readRDS("tumor_sample.rds")
# Run analysis with calibrated thresholds
result <- scPharmIdentify(
tumor_seurat,
type = "tissue",
cancer = "LUAD",
drug = "Erlotinib",
threshold.s = threshold_s,
threshold.r = threshold_r
)Threshold Selection Strategy
| Scenario | Recommendation |
|---|---|
| Strict classification | Higher threshold.s, lower threshold.r
|
| Lenient classification | Lower threshold.s, higher threshold.r
|
| Balanced | Use scPharmGenNullDist() defaults |
2. Multi-Drug Analysis
Pan-Drug Screening
# Analyze all available drugs
result_all <- scPharmIdentify(
seurat_obj,
type = "cellline",
cancer = "BRCA",
drug = "all",
cores = 8 # Parallel processing
)
# Get drug ranking
dr_scores <- scPharmDr(result_all)
print(head(dr_scores, 20))Drug Class Analysis
# Load drug metadata
data(drug_info, package = "scPharm")
# Filter by drug class
chemo_drugs <- drug_info %>%
filter(DRUG_TYPE == "chemotherapy") %>%
pull(DRUG_NAME)
targeted_drugs <- drug_info %>%
filter(DRUG_TYPE == "targeted") %>%
pull(DRUG_NAME)
# Analyze specific drug classes
result_chemo <- scPharmIdentify(
seurat_obj,
cancer = "BRCA",
drug = chemo_drugs
)
result_targeted <- scPharmIdentify(
seurat_obj,
cancer = "BRCA",
drug = targeted_drugs
)Multi-Cancer Analysis
# Analyze across multiple cancer contexts
cancers <- c("BRCA", "LUAD", "COREAD")
results <- lapply(cancers, function(cancer) {
result <- scPharmIdentify(
seurat_obj,
type = "cellline",
cancer = cancer,
drug = "Paclitaxel"
)
# Extract NES values
nes_col <- grep("scPharm_nes_", colnames(result@meta.data), value = TRUE)
data.frame(
cancer = cancer,
mean_nes = mean(result@meta.data[[nes_col]], na.rm = TRUE),
sd_nes = sd(result@meta.data[[nes_col]], na.rm = TRUE)
)
})
# Combine results
multi_cancer_df <- do.call(rbind, results)
print(multi_cancer_df)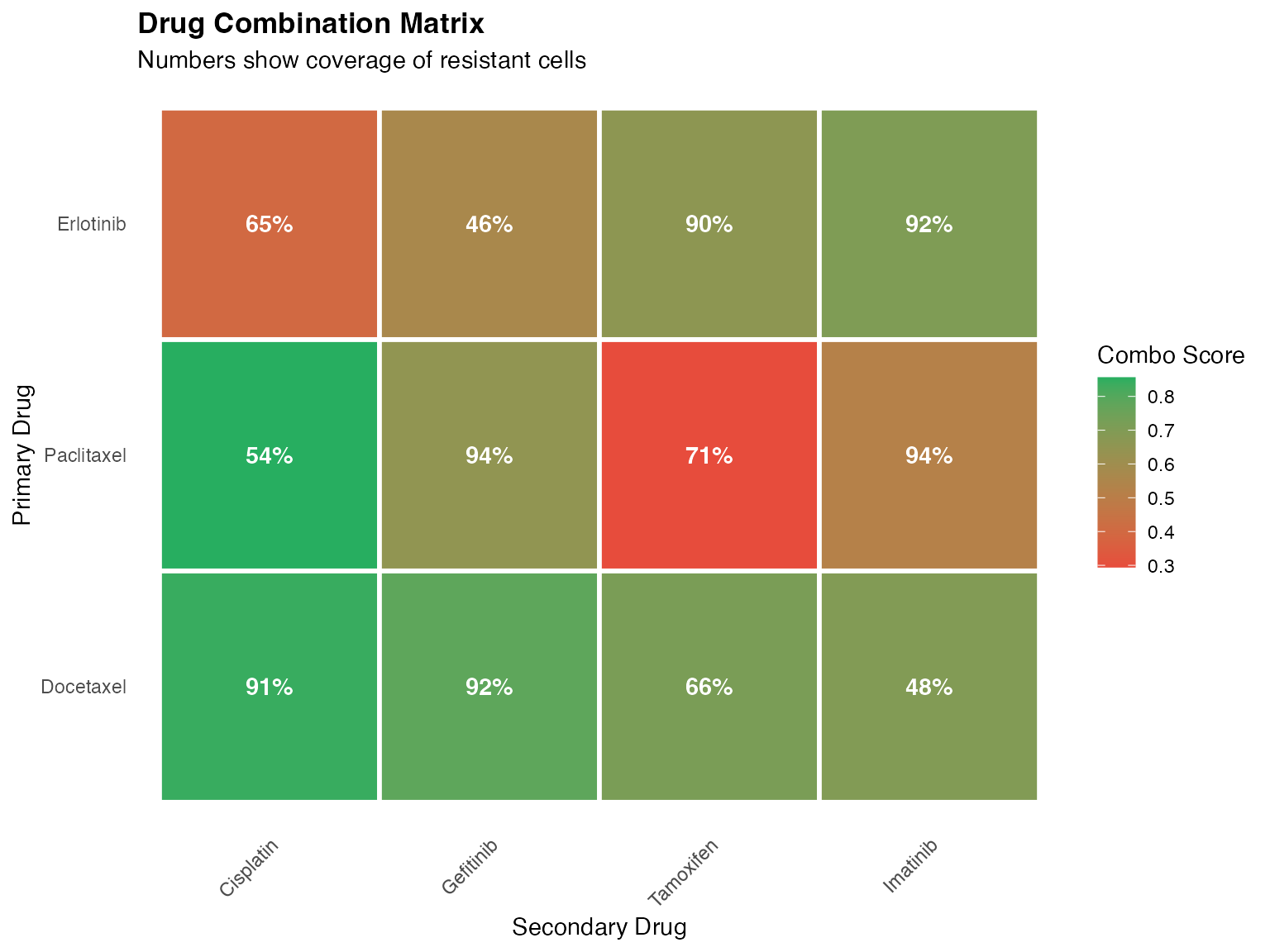
4. Integration with Seurat Workflows
Pre-computed Embeddings
# Use existing Seurat clustering
seurat_obj <- FindVariableFeatures(seurat_obj)
seurat_obj <- ScaleData(seurat_obj)
seurat_obj <- RunPCA(seurat_obj)
seurat_obj <- FindNeighbors(seurat_obj)
seurat_obj <- FindClusters(seurat_obj)
seurat_obj <- RunUMAP(seurat_obj, dims = 1:30)
# Run scPharm (MCA is computed independently)
result <- scPharmIdentify(seurat_obj, ...)
# Visualize with Seurat functions
DimPlot(result, group.by = "scPharm_label_Docetaxel")
FeaturePlot(result, features = "scPharm_nes_Docetaxel")Cluster-Level Analysis
# Aggregate NES by cluster
cluster_summary <- result@meta.data %>%
group_by(seurat_clusters) %>%
summarise(
n_cells = n(),
mean_nes = mean(scPharm_nes_Docetaxel, na.rm = TRUE),
pct_sensitive = mean(scPharm_label_Docetaxel == "sensitive") * 100,
pct_resistant = mean(scPharm_label_Docetaxel == "resistant") * 100
)
print(cluster_summary)5. Performance Tuning
Memory Management
For large datasets, consider:
# Process in chunks
cell_chunks <- split(colnames(seurat_obj),
ceiling(seq_along(colnames(seurat_obj)) / 5000))
results <- lapply(cell_chunks, function(cells) {
subset_obj <- subset(seurat_obj, cells = cells)
scPharmIdentify(subset_obj, ...)
})
# Merge results
final_result <- merge(results[[1]], results[-1])Parallel Processing
# Detect available cores
n_cores <- parallel::detectCores() - 1
# Run with multiple cores
result <- scPharmIdentify(
seurat_obj,
drug = "all",
cores = n_cores
)7. Quality Control
Pre-analysis Checks
# Check gene coverage
data(bulkdata, package = "scPharm")
gene_overlap <- intersect(rownames(seurat_obj), rownames(bulkdata))
cat("Gene overlap:", length(gene_overlap), "/", nrow(bulkdata), "\n")
# Minimum recommended: 5000 genes
if (length(gene_overlap) < 5000) {
warning("Low gene overlap may affect accuracy")
}
# Check cell numbers
if (ncol(seurat_obj) < 100) {
warning("Small cell numbers may lead to unstable estimates")
}8. Troubleshooting
Common Issues
| Issue | Possible Cause | Solution |
|---|---|---|
| All cells classified as “other” | Thresholds too strict | Lower threshold_s, raise threshold_r |
| No tumor cells detected | CNV detection failed | Provide tumor.cells manually |
| Memory error | Dataset too large | Process in chunks |
| Slow performance | Too many drugs | Use parallel processing |
Debug Mode
# Verbose output
options(scPharm.verbose = TRUE)
# Step-by-step execution
result <- scPharmIdentify(
seurat_obj,
type = "cellline",
cancer = "BRCA",
drug = "Docetaxel"
)9. Exporting Results
To Seurat Object
# Save annotated Seurat object
saveRDS(result, "annotated_seurat.rds")Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] dplyr_1.1.4 ggplot2_4.0.1
#>
#> loaded via a namespace (and not attached):
#> [1] gtable_0.3.6 jsonlite_2.0.0 compiler_4.4.0 tidyselect_1.2.1
#> [5] dichromat_2.0-0.1 jquerylib_0.1.4 systemfonts_1.3.1 scales_1.4.0
#> [9] textshaping_1.0.4 yaml_2.3.12 fastmap_1.2.0 R6_2.6.1
#> [13] labeling_0.4.3 generics_0.1.4 knitr_1.51 htmlwidgets_1.6.4
#> [17] tibble_3.3.1 desc_1.4.3 bslib_0.9.0 pillar_1.11.1
#> [21] RColorBrewer_1.1-3 rlang_1.1.7 cachem_1.1.0 xfun_0.56
#> [25] fs_1.6.6 sass_0.4.10 S7_0.2.1 otel_0.2.0
#> [29] cli_3.6.5 pkgdown_2.1.3 withr_3.0.2 magrittr_2.0.4
#> [33] digest_0.6.39 grid_4.4.0 lifecycle_1.0.5 vctrs_0.7.1
#> [37] evaluate_1.0.5 glue_1.8.0 farver_2.1.2 ragg_1.5.0
#> [41] rmarkdown_2.30 tools_4.4.0 pkgconfig_2.0.3 htmltools_0.5.9