Introduction
This vignette covers advanced features of darwin for power users, including custom objective functions, fixed gene count mode, parallel computing, and integration with other tools.
Prepare Data
# Create reference matrix
n_ct <- 5
n_genes <- 400
reference <- matrix(abs(rnorm(n_ct * n_genes, 2, 1)), nrow = n_ct, ncol = n_genes)
rownames(reference) <- paste0("CellType", 1:n_ct)
colnames(reference) <- paste0("Gene", 1:n_genes)
# Add markers
for (i in 1:n_ct) {
reference[i, ((i-1)*20+1):(i*20)] <- reference[i, ((i-1)*20+1):(i*20)] + 4
}Custom Objective Functions
darwin allows you to define custom objective functions for specialized applications.
Requirements
A valid objective function must:
- Accept a single argument: data matrix (cell types × genes)
- Return a single numeric value
- Be consistent (same input → same output)
Example: Marker Score Objective
# Custom objective: maximize marker specificity
# Higher when genes are specific to individual cell types
marker_score <- function(data) {
# Max expression / mean expression ratio
col_max <- apply(data, 2, max)
col_mean <- colMeans(data)
col_mean[col_mean == 0] <- 1e-10
sum(col_max / col_mean)
}
cat("Marker score value:", marker_score(test_data), "\n")
#> Marker score value: 107.8375Using Custom Objectives
dw_custom <- darwin(reference)
dw_custom$optimize(
ngen = 60,
objectives = c("correlation", variance_objective),
weights = c(-1, 1), # Minimize correlation, maximize variance
verbose = FALSE,
parallel = FALSE
)
# The second objective column will show variance values
fitness <- dw_custom$get_fitness()
colnames(fitness) <- c("Correlation", "Variance")
ggplot(as.data.frame(fitness), aes(x = Correlation, y = Variance)) +
geom_point(color = "#3498db", size = 3, alpha = 0.7) +
geom_line(color = "gray60", alpha = 0.5) +
labs(
title = "Custom Objectives: Correlation vs Variance",
subtitle = "Minimize correlation, maximize variance",
x = "Correlation (lower is better)",
y = "Variance (higher is better)"
) +
theme_minimal(base_size = 12)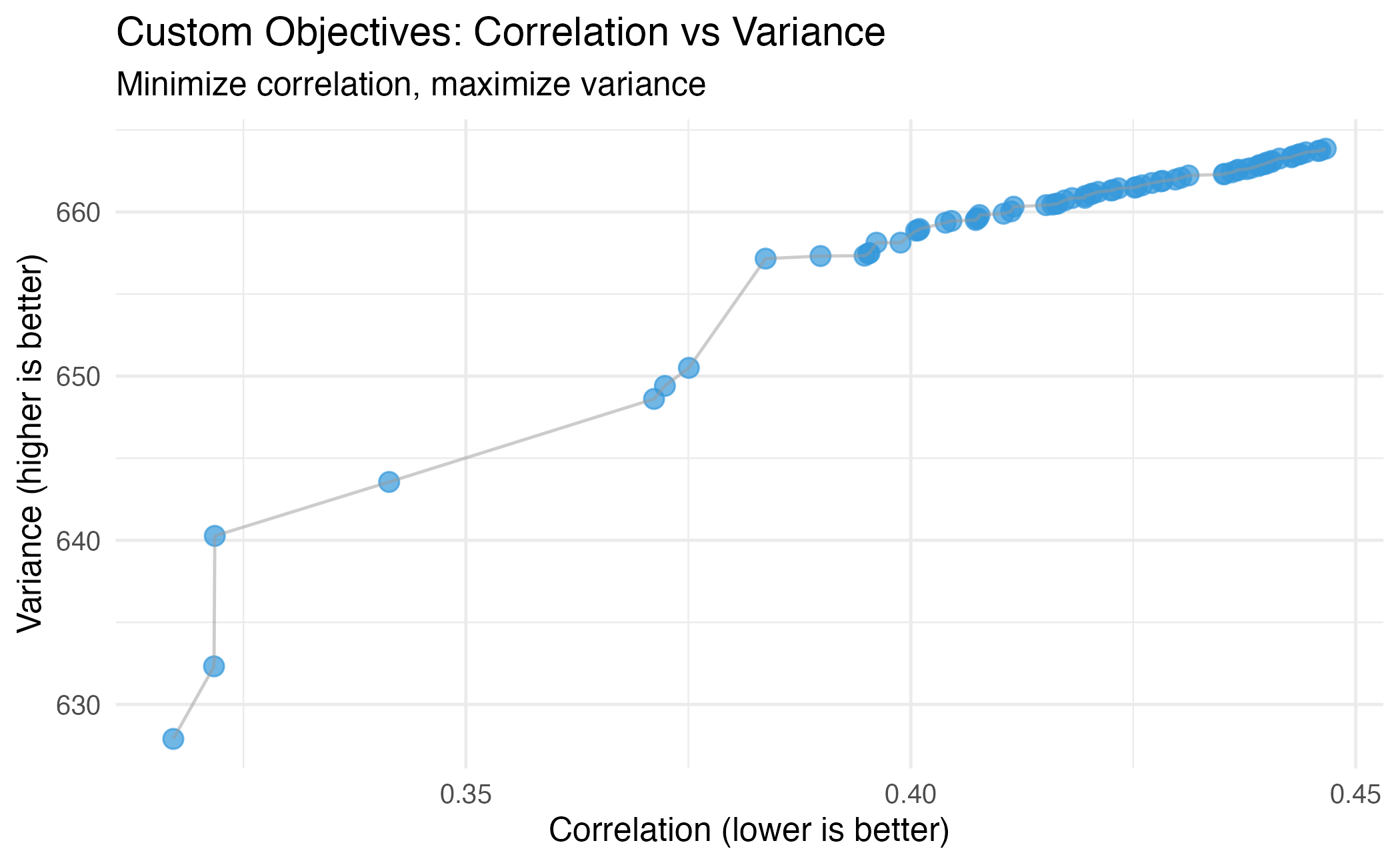
Pareto front with custom objectives: correlation vs variance.
Three Objectives
dw_three <- darwin(reference)
dw_three$optimize(
ngen = 60,
objectives = c("correlation", "distance", "condition"),
weights = c(-1, 1, -1), # Minimize corr/cond, maximize distance
verbose = FALSE,
parallel = FALSE
)
fitness3 <- dw_three$get_fitness()
cat("Three-objective optimization:\n")
#> Three-objective optimization:
cat(" Solutions:", nrow(fitness3), "\n")
#> Solutions: 314
cat(" Correlation range:", round(range(fitness3[,1]), 2), "\n")
#> Correlation range: 0.21 0.79
cat(" Distance range:", round(range(fitness3[,2]), 2), "\n")
#> Distance range: 143.43 363.86
cat(" Condition range:", round(range(fitness3[,3]), 2), "\n")
#> Condition range: 3.93 4.27Fixed Gene Count Mode
For applications requiring a specific number of marker genes, use fixed mode:
dw_fixed <- darwin(reference)
dw_fixed$optimize(
ngen = 60,
mode = "fixed",
n_features = 50, # Select exactly 50 genes
objectives = c("correlation", "distance"),
weights = c(-1, 1),
verbose = FALSE,
parallel = FALSE
)
# Verify all solutions have 50 genes
pareto <- dw_fixed$get_pareto()
gene_counts <- sapply(pareto, sum)
cat("Gene counts in fixed mode:", unique(gene_counts), "\n")
#> Gene counts in fixed mode: 50
# Compare gene count distributions
df_compare <- rbind(
data.frame(
mode = "Standard",
n_genes = sapply(dw_custom$get_pareto(), sum)
),
data.frame(
mode = "Fixed (50)",
n_genes = gene_counts
)
)
ggplot(df_compare, aes(x = n_genes, fill = mode)) +
geom_histogram(bins = 20, alpha = 0.7, position = "identity") +
scale_fill_manual(values = c("#3498db", "#e74c3c")) +
labs(
title = "Gene Count Distribution: Standard vs Fixed Mode",
x = "Number of Selected Genes",
y = "Frequency"
) +
theme_minimal(base_size = 12)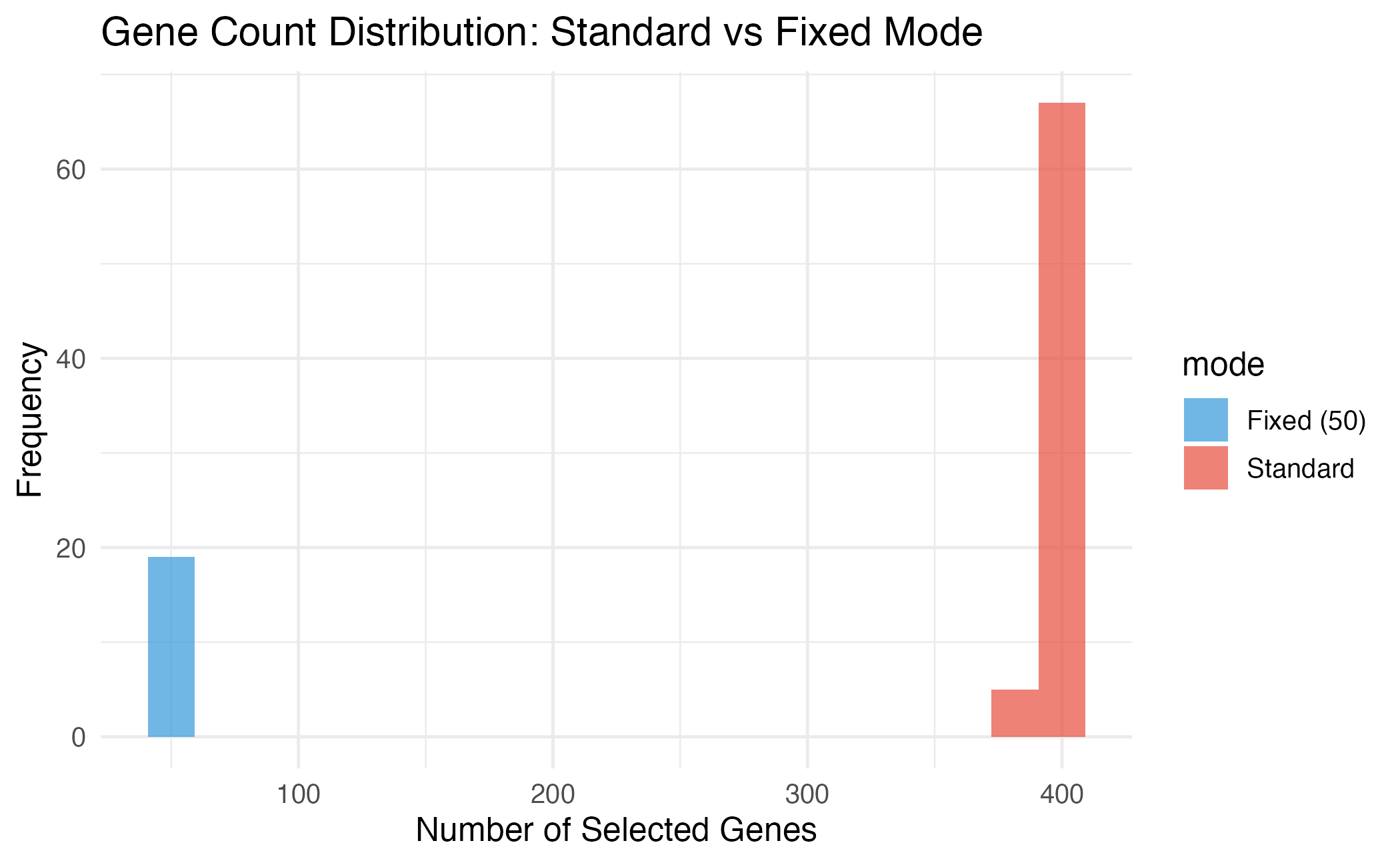
Fixed mode ensures all solutions have exactly the specified number of genes.
Selection Strategies
darwin provides flexible selection from the Pareto front:
1. Weighted Selection
dw <- darwin(reference)
dw$optimize(ngen = 50, verbose = FALSE, parallel = FALSE)
# Emphasize minimizing correlation
dw$select(weights = c(-2, 1))
cat("Emphasize correlation - genes:", sum(dw$get_selection()), "\n")
#> Emphasize correlation - genes: 388
# Emphasize maximizing distance
dw$select(weights = c(-1, 2))
cat("Emphasize distance - genes:", sum(dw$get_selection()), "\n")
#> Emphasize distance - genes: 3882. Index-Based Selection
# Select by direct index
dw$select(index = 1)
cat("Solution 1 - genes:", sum(dw$get_selection()), "\n")
#> Solution 1 - genes: 400
# Select by objective rank
# (objective_index, rank) - rank 1 = best for that objective
dw$select(index = c(1, 1)) # Best correlation
cat("Best correlation - genes:", sum(dw$get_selection()), "\n")
#> Best correlation - genes: 326
dw$select(index = c(2, -1)) # Best distance (last rank)
cat("Best distance - genes:", sum(dw$get_selection()), "\n")
#> Best distance - genes: 400Parallel Computing
darwin supports parallel computation for faster optimization:
# Enable parallel computing
options(darwin.parallel = TRUE)
# Or specify in optimize()
dw$optimize(
ngen = 100,
parallel = TRUE, # Uses all available cores - 1
verbose = TRUE
)
# Disable parallel computing
options(darwin.parallel = FALSE)Performance Comparison
# Benchmark (example, not run)
library(microbenchmark)
# Large dataset
large_ref <- matrix(rnorm(50 * 2000), nrow = 50)
microbenchmark(
serial = {
dw <- darwin(large_ref)
dw$optimize(ngen = 10, parallel = FALSE, verbose = FALSE)
},
parallel = {
dw <- darwin(large_ref)
dw$optimize(ngen = 10, parallel = TRUE, verbose = FALSE)
},
times = 3
)Parameter Tuning
Population Size
Larger populations provide more diversity but slower convergence:
results <- list()
for (pop in c(30, 60, 100)) {
dw_temp <- darwin(reference)
dw_temp$optimize(
ngen = 40,
pop_size = pop,
verbose = FALSE,
parallel = FALSE
)
results[[as.character(pop)]] <- data.frame(
dw_temp$get_fitness(),
pop_size = factor(pop)
)
}
df_pop <- do.call(rbind, results)
ggplot(df_pop, aes(x = correlation, y = distance, color = pop_size)) +
geom_point(alpha = 0.6) +
scale_color_brewer(palette = "Set1") +
labs(
title = "Effect of Population Size",
x = "Correlation",
y = "Distance",
color = "Population\nSize"
) +
theme_minimal(base_size = 12)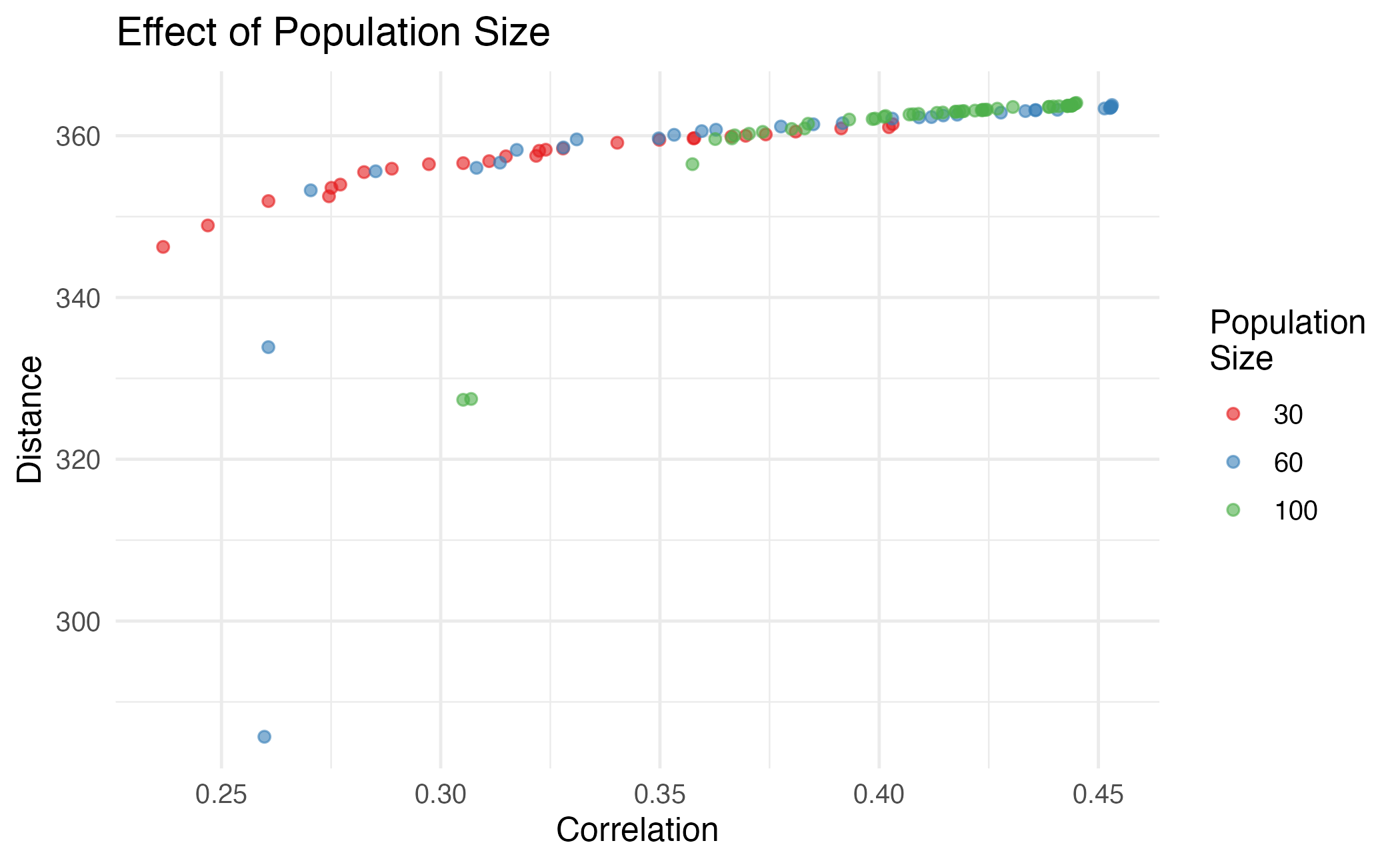
Effect of population size on Pareto front diversity.
Mutation Probability
Higher mutation enables more exploration:
results_mut <- list()
for (mut in c(0.001, 0.01, 0.05)) {
dw_temp <- darwin(reference)
dw_temp$optimize(
ngen = 40,
mutation_prob = mut,
verbose = FALSE,
parallel = FALSE
)
pareto <- dw_temp$get_pareto()
gene_counts <- sapply(pareto, sum)
results_mut[[as.character(mut)]] <- data.frame(
n_genes = gene_counts,
mutation = factor(mut)
)
}
df_mut <- do.call(rbind, results_mut)
ggplot(df_mut, aes(x = n_genes, fill = mutation)) +
geom_density(alpha = 0.5) +
scale_fill_brewer(palette = "Set1") +
labs(
title = "Effect of Mutation Probability on Gene Count",
x = "Number of Selected Genes",
y = "Density",
fill = "Mutation\nProbability"
) +
theme_minimal(base_size = 12)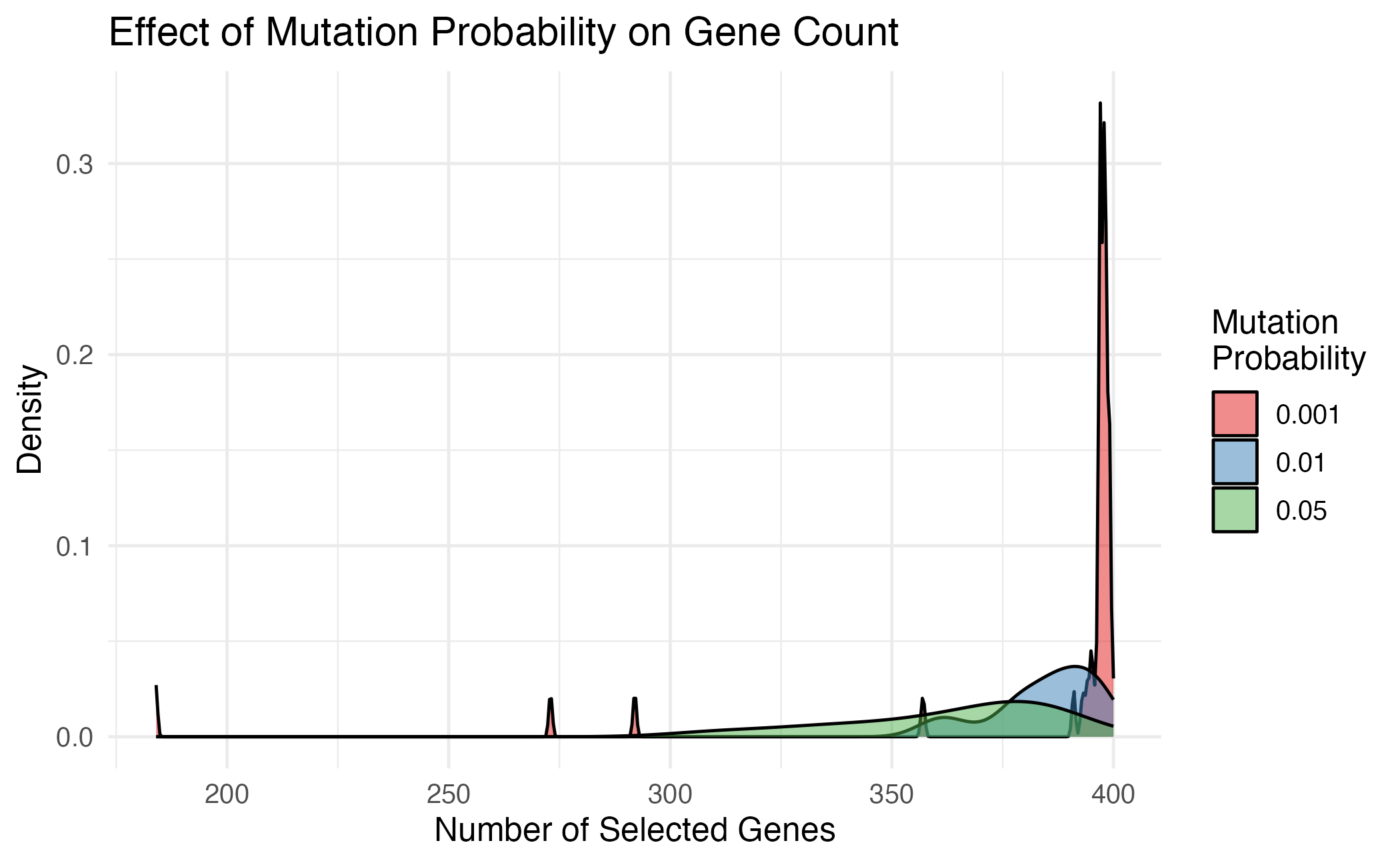
Effect of mutation probability on solution diversity.
Save and Load
darwin objects can be saved and loaded for reproducibility:
# Save
temp_file <- tempfile(fileext = ".rds")
dw$save(temp_file)
# Load
dw_loaded <- readRDS(temp_file)
# Verify
cat("Original Pareto size:", length(dw$get_pareto()), "\n")
#> Original Pareto size: 99
cat("Loaded Pareto size:", length(dw_loaded$get_pareto()), "\n")
#> Loaded Pareto size: 99
# Clean up
unlink(temp_file)Integration Examples
Export to Other Deconvolution Tools
# Get selected genes for use with other tools
genes <- dw$get_genes()
selection <- dw$get_selection()
# Create signature matrix for CIBERSORT
signature_matrix <- reference[, selection]
write.csv(signature_matrix, "signature_matrix.csv")
# Or as gene list for MuSiC
gene_list <- genesTroubleshooting
Common Issues
-
Slow optimization: Reduce
ngen,pop_size, or enableparallel = TRUE -
Poor convergence: Increase
ngenorpop_size -
All solutions identical: Increase
mutation_prob -
Memory issues: Use gene pre-selection with
use_highly_variable = TRUE
Diagnostic Checks
# Check optimization quality
fitness <- dw$get_fitness()
cat("Diagnostic Summary:\n")
#> Diagnostic Summary:
cat(" Pareto front size:", nrow(fitness), "\n")
#> Pareto front size: 99
cat(" Correlation range:", round(diff(range(fitness[,1])), 3), "\n")
#> Correlation range: 0.149
cat(" Distance range:", round(diff(range(fitness[,2])), 3), "\n")
#> Distance range: 47.774
cat(" Gene count range:", range(sapply(dw$get_pareto(), sum)), "\n")
#> Gene count range: 326 400Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggplot2_4.0.1 darwin_1.0.0
#>
#> loaded via a namespace (and not attached):
#> [1] sass_0.4.10 future_1.69.0 generics_0.1.4
#> [4] lattice_0.22-7 listenv_0.10.0 digest_0.6.39
#> [7] magrittr_2.0.4 evaluate_1.0.5 grid_4.4.0
#> [10] RColorBrewer_1.1-3 fastmap_1.2.0 jsonlite_2.0.0
#> [13] Matrix_1.7-4 scales_1.4.0 codetools_0.2-20
#> [16] textshaping_1.0.4 jquerylib_0.1.4 cli_3.6.5
#> [19] rlang_1.1.7 parallelly_1.46.1 future.apply_1.20.1
#> [22] withr_3.0.2 cachem_1.1.0 yaml_2.3.12
#> [25] otel_0.2.0 tools_4.4.0 parallel_4.4.0
#> [28] dplyr_1.1.4 globals_0.18.0 vctrs_0.7.1
#> [31] R6_2.6.1 lifecycle_1.0.5 fs_1.6.6
#> [34] htmlwidgets_1.6.4 ragg_1.5.0 pkgconfig_2.0.3
#> [37] desc_1.4.3 pkgdown_2.1.3 pillar_1.11.1
#> [40] bslib_0.9.0 gtable_0.3.6 glue_1.8.0
#> [43] Rcpp_1.1.1 systemfonts_1.3.1 xfun_0.56
#> [46] tibble_3.3.1 tidyselect_1.2.1 knitr_1.51
#> [49] dichromat_2.0-0.1 farver_2.1.2 htmltools_0.5.9
#> [52] rmarkdown_2.30 labeling_0.4.3 compiler_4.4.0
#> [55] S7_0.2.1