Algorithm Theory: NSGA-II for Gene Selection
Zaoqu Liu
2026-01-25
Source:vignettes/algorithm.Rmd
algorithm.RmdIntroduction
This vignette provides a detailed explanation of the algorithms and mathematical foundations underlying darwin. Understanding these concepts will help users make informed decisions about parameter settings and interpret results correctly.
The Multi-Objective Optimization Problem
Problem Formulation
Given a reference expression matrix where:
- = number of cell types
- = number of genes
We seek a gene subset that optimizes multiple conflicting objectives simultaneously:
where is the submatrix of containing only the genes in .
Pareto Dominance
In multi-objective optimization, we use the concept of Pareto dominance to compare solutions:
Definition: Solution dominates solution (written ) if and only if:
- is no worse than in all objectives:
- is strictly better than in at least one objective:
Pareto Front
The Pareto front (or Pareto-optimal set) consists of all non-dominated solutions—solutions that cannot be improved in any objective without worsening another.
library(darwin)
library(ggplot2)
# Create example fitness landscape
set.seed(42)
n_points <- 100
f1 <- runif(n_points, 0, 10)
f2 <- 10 - f1 + rnorm(n_points, sd = 2)
f2[f2 < 0] <- 0.5
# Find Pareto front
is_pareto <- rep(FALSE, n_points)
for (i in 1:n_points) {
dominated <- FALSE
for (j in 1:n_points) {
if (i != j && f1[j] <= f1[i] && f2[j] >= f2[i] && (f1[j] < f1[i] || f2[j] > f2[i])) {
dominated <- TRUE
break
}
}
is_pareto[i] <- !dominated
}
df <- data.frame(correlation = f1, distance = f2, pareto = is_pareto)
ggplot(df, aes(x = correlation, y = distance, color = pareto)) +
geom_point(size = 3, alpha = 0.7) +
scale_color_manual(values = c("gray60", "#3498db"), labels = c("Dominated", "Pareto Front")) +
geom_line(data = df[df$pareto, ][order(df[df$pareto, ]$correlation), ],
color = "#3498db", linewidth = 1) +
labs(
title = "Pareto Front Visualization",
subtitle = "Trade-off between minimizing correlation and maximizing distance",
x = "Correlation (minimize)",
y = "Distance (maximize)",
color = "Solution Type"
) +
theme_minimal(base_size = 12) +
theme(legend.position = "bottom")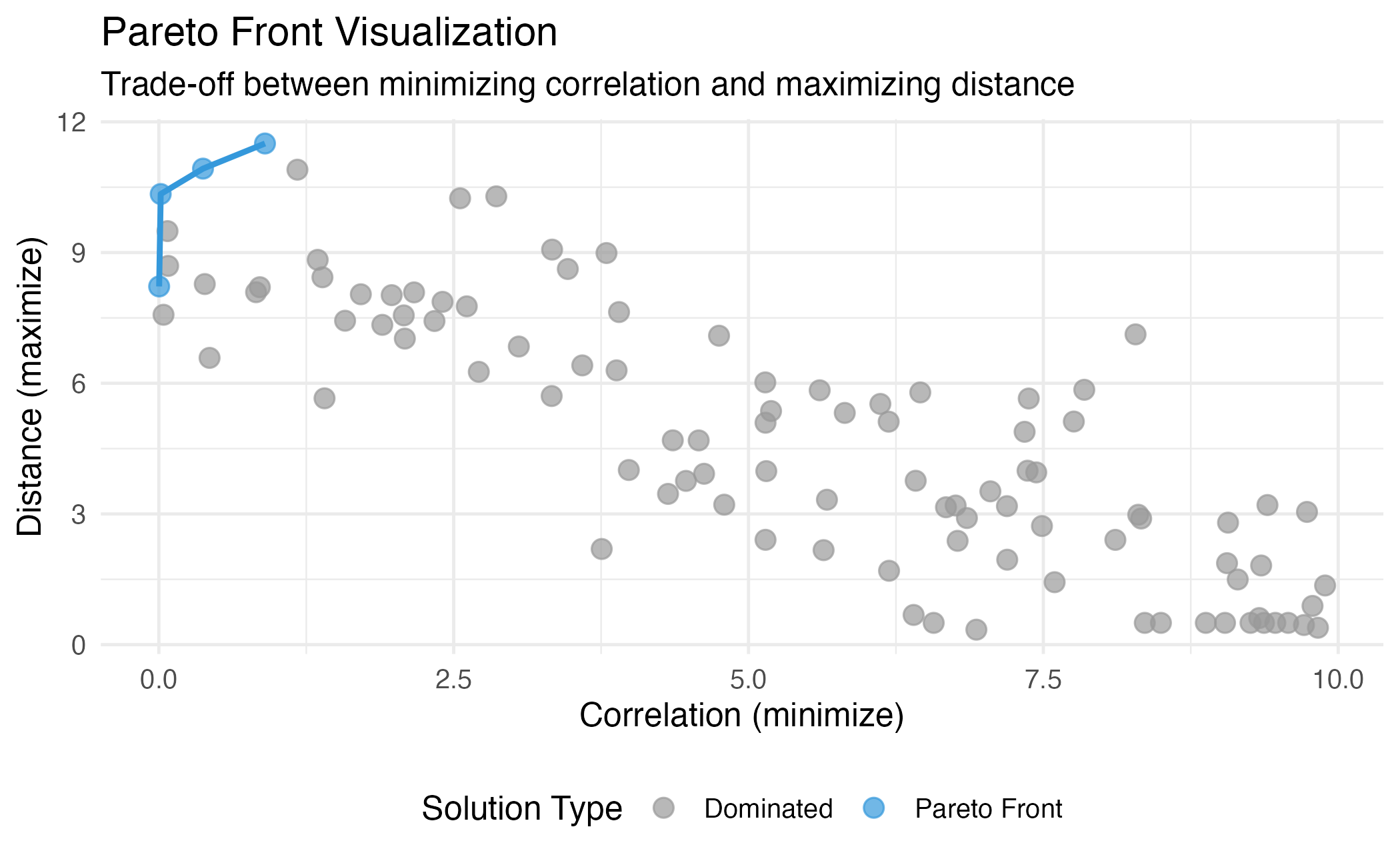
Illustration of Pareto dominance and the Pareto front. Points on the front (blue) are non-dominated.
NSGA-II Algorithm
darwin implements NSGA-II (Non-dominated Sorting Genetic Algorithm II), a widely-used evolutionary algorithm for multi-objective optimization.
Algorithm Overview
Algorithm: NSGA-II
Input: Population size N, generations G, objectives f₁...fₘ
Output: Pareto-optimal gene subsets
1. Initialize population P₀ with N random gene selections
2. Evaluate objectives for all individuals
3. For generation t = 1 to G:
a. Create offspring Q_t using selection, crossover, mutation
b. Combine: R_t = P_t ∪ Q_t
c. Non-dominated sorting of R_t into fronts F₁, F₂, ...
d. Select N individuals for P_{t+1} using fronts and crowding distance
4. Return final Pareto frontKey Components
1. Non-dominated Sorting
Individuals are sorted into fronts based on dominance:
- Front 1: All non-dominated individuals
- Front 2: Non-dominated among remaining individuals
- Front k: Continue until all individuals are assigned
# Simulate fitness values
set.seed(123)
n <- 20
fitness <- matrix(runif(n * 2), ncol = 2)
colnames(fitness) <- c("Obj1", "Obj2")
# Non-dominated sorting (simplified)
ranks <- darwin:::.non_dominated_sort(-fitness) # Negate for maximization
df_fronts <- data.frame(
Obj1 = fitness[, 1],
Obj2 = fitness[, 2],
Front = factor(ranks)
)
ggplot(df_fronts, aes(x = Obj1, y = Obj2, color = Front, shape = Front)) +
geom_point(size = 4) +
scale_color_brewer(palette = "Set1") +
labs(
title = "Non-dominated Sorting",
subtitle = "Individuals assigned to Pareto fronts",
x = "Objective 1",
y = "Objective 2"
) +
theme_minimal(base_size = 12)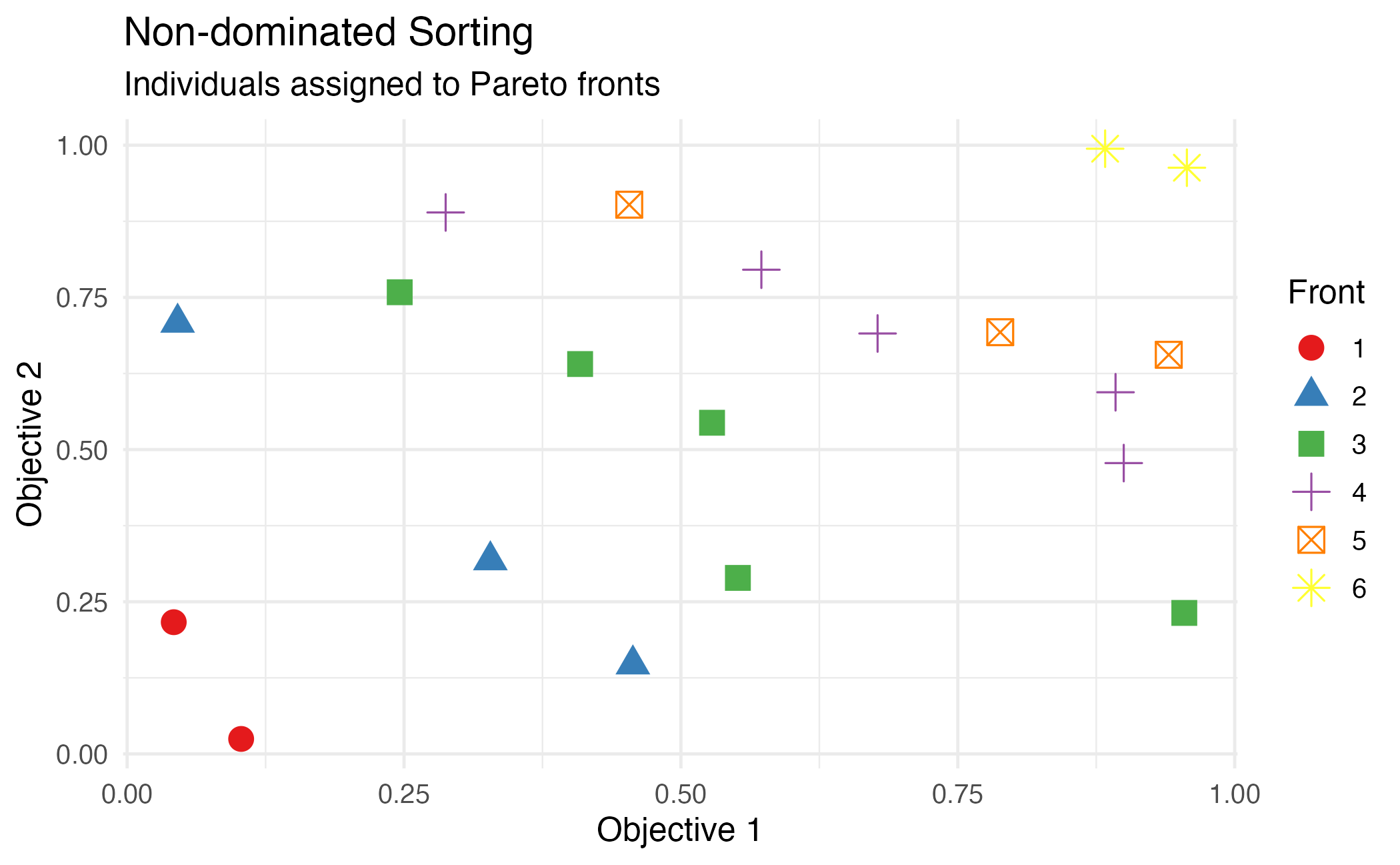
Non-dominated sorting assigns individuals to Pareto fronts.
2. Crowding Distance
Within each front, crowding distance measures the density of solutions around each individual. Higher crowding distance indicates more isolated solutions, which are preferred to maintain diversity.
For individual in front :
where and are the objective values of neighboring individuals when sorted by objective .
set.seed(456)
n <- 15
f1 <- sort(runif(n, 1, 10))
f2 <- 10 - f1 + rnorm(n, sd = 0.3)
fitness_mat <- cbind(f1, f2)
crowding <- crowding_distance(fitness_mat, ranks = rep(1, n))
crowding[is.infinite(crowding)] <- max(crowding[is.finite(crowding)]) * 1.5
df_crowd <- data.frame(f1 = f1, f2 = f2, crowding = crowding)
ggplot(df_crowd, aes(x = f1, y = f2, size = crowding, color = crowding)) +
geom_point(alpha = 0.8) +
scale_size_continuous(range = c(3, 10)) +
scale_color_gradient(low = "#3498db", high = "#e74c3c") +
labs(
title = "Crowding Distance Visualization",
subtitle = "Larger points = higher crowding distance (more isolated)",
x = "Objective 1",
y = "Objective 2",
size = "Crowding\nDistance",
color = "Crowding\nDistance"
) +
theme_minimal(base_size = 12)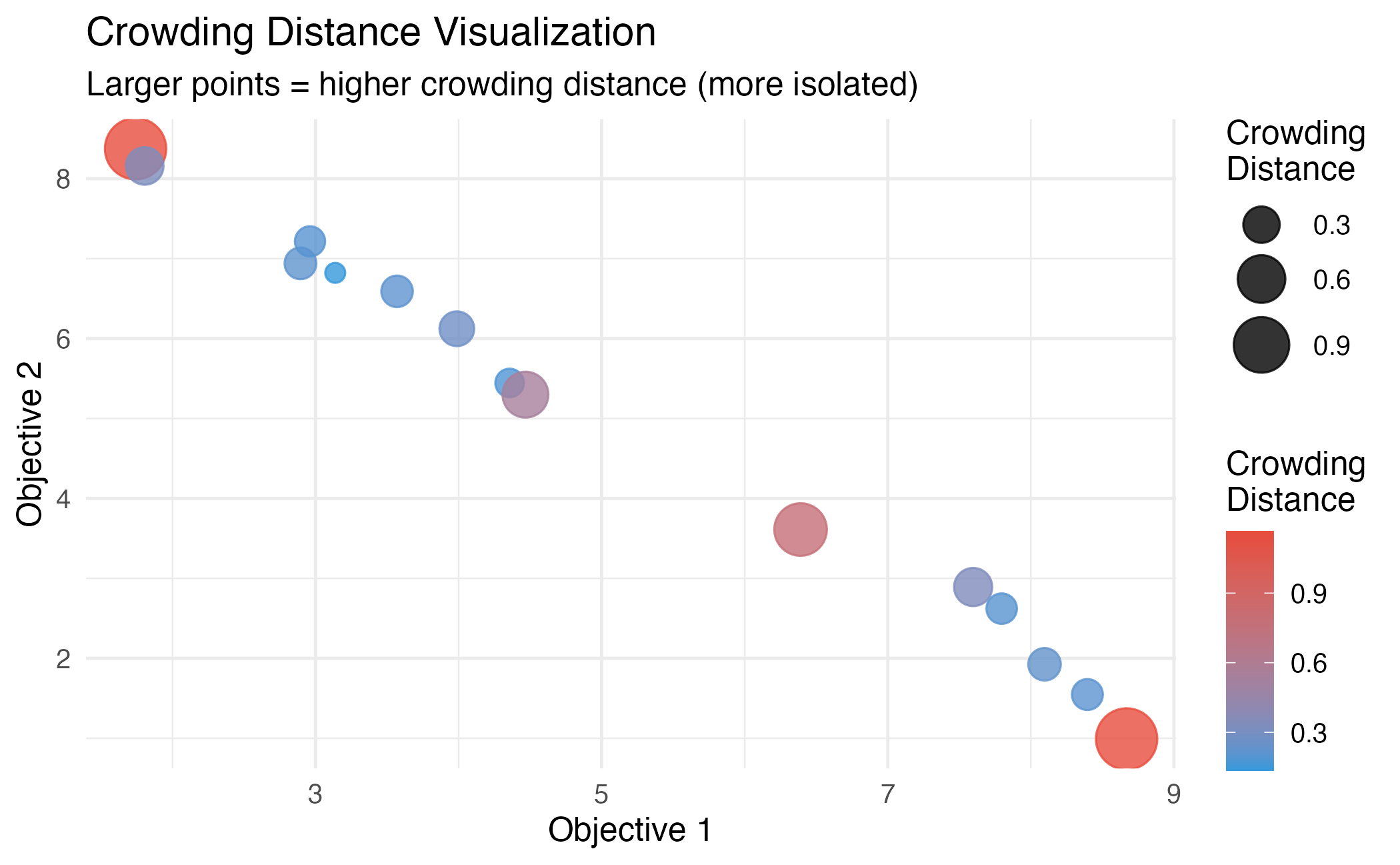
Crowding distance computation. Boundary solutions receive infinite distance.
Objective Functions
Correlation Objective (Minimize)
Measures the total pairwise correlation between cell type expression profiles:
where is the Pearson correlation coefficient between cell types and .
Intuition: Low correlation ensures cell types are distinguishable, improving deconvolution accuracy.
library(darwin)
set.seed(42)
# Create data with structure
n_ct <- 5
n_genes <- 100
data <- matrix(rnorm(n_ct * n_genes), nrow = n_ct)
rownames(data) <- paste0("CT", 1:n_ct)
# Add markers
for (i in 1:n_ct) {
data[i, ((i-1)*10+1):(i*10)] <- data[i, ((i-1)*10+1):(i*10)] + 3
}
# Compute correlation
corr_mat <- cor(t(data))
# Plot
df_corr <- expand.grid(CT1 = rownames(corr_mat), CT2 = colnames(corr_mat))
df_corr$correlation <- as.vector(corr_mat)
ggplot(df_corr, aes(x = CT1, y = CT2, fill = correlation)) +
geom_tile() +
scale_fill_gradient2(low = "#3498db", mid = "white", high = "#e74c3c", midpoint = 0) +
labs(
title = "Cell Type Correlation Matrix",
subtitle = paste("Total absolute correlation:", round(compute_correlation(data), 2)),
x = "", y = ""
) +
theme_minimal(base_size = 12) +
coord_fixed()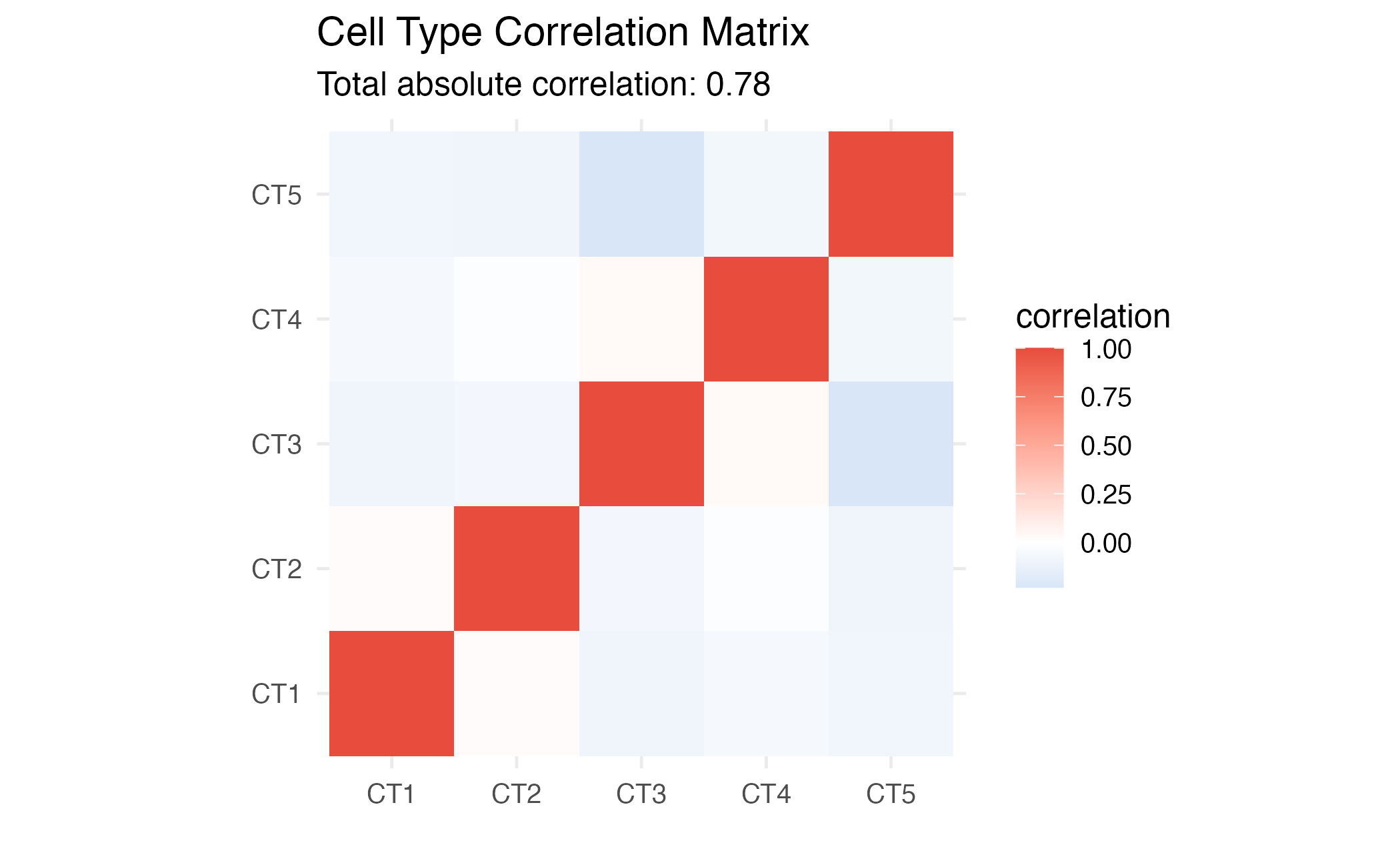
Correlation matrix heatmap for selected genes.
Parameter Guidelines
| Parameter | Typical Range | Effect |
|---|---|---|
ngen |
50-500 | More generations = better convergence, longer runtime |
pop_size |
50-200 | Larger = more diversity, slower |
mutation_prob |
0.001-0.01 | Higher = more exploration |
crossover_prob |
0.6-0.9 | Higher = more recombination |
References
NSGA-II: Deb, K., Pratap, A., Agarwal, S., & Meyarivan, T. (2002). A fast and elitist multiobjective genetic algorithm: NSGA-II. IEEE Transactions on Evolutionary Computation, 6(2), 182-197.
AutoGeneS: Aliee, H., & Theis, F. J. (2021). AutoGeneS: Automatic gene selection using multi-objective optimization for RNA-seq deconvolution. Cell Systems, 12(7), 706-715.e4.
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggplot2_4.0.1 darwin_1.0.0
#>
#> loaded via a namespace (and not attached):
#> [1] sass_0.4.10 future_1.69.0 generics_0.1.4
#> [4] lattice_0.22-7 listenv_0.10.0 digest_0.6.39
#> [7] magrittr_2.0.4 evaluate_1.0.5 grid_4.4.0
#> [10] RColorBrewer_1.1-3 fastmap_1.2.0 jsonlite_2.0.0
#> [13] Matrix_1.7-4 scales_1.4.0 codetools_0.2-20
#> [16] textshaping_1.0.4 jquerylib_0.1.4 cli_3.6.5
#> [19] rlang_1.1.7 parallelly_1.46.1 future.apply_1.20.1
#> [22] withr_3.0.2 cachem_1.1.0 yaml_2.3.12
#> [25] otel_0.2.0 tools_4.4.0 parallel_4.4.0
#> [28] dplyr_1.1.4 globals_0.18.0 vctrs_0.7.1
#> [31] R6_2.6.1 lifecycle_1.0.5 fs_1.6.6
#> [34] htmlwidgets_1.6.4 ragg_1.5.0 pkgconfig_2.0.3
#> [37] desc_1.4.3 pkgdown_2.1.3 pillar_1.11.1
#> [40] bslib_0.9.0 gtable_0.3.6 glue_1.8.0
#> [43] Rcpp_1.1.1 systemfonts_1.3.1 xfun_0.56
#> [46] tibble_3.3.1 tidyselect_1.2.1 knitr_1.51
#> [49] dichromat_2.0-0.1 farver_2.1.2 htmltools_0.5.9
#> [52] rmarkdown_2.30 labeling_0.4.3 compiler_4.4.0
#> [55] S7_0.2.1