Introduction
This vignette demonstrates various visualization techniques for analyzing darwin optimization results. Effective visualization is crucial for understanding the trade-offs in multi-objective optimization and selecting appropriate solutions.
Prepare Example Data
# Create reference expression matrix
n_celltypes <- 6
n_genes <- 300
reference <- matrix(
abs(rnorm(n_celltypes * n_genes, mean = 2)),
nrow = n_celltypes,
ncol = n_genes
)
rownames(reference) <- c("B_cells", "T_cells", "NK_cells", "Monocytes", "Dendritic", "Neutrophils")
colnames(reference) <- paste0("Gene", 1:n_genes)
# Add cell-type specific markers
for (i in 1:n_celltypes) {
markers <- ((i - 1) * 15 + 1):(i * 15)
reference[i, markers] <- reference[i, markers] + runif(15, 3, 6)
}
# Run optimization
dw <- darwin(reference)
dw$optimize(
ngen = 80,
pop_size = 80,
objectives = c("correlation", "distance"),
weights = c(-1, 1),
verbose = FALSE,
parallel = FALSE
)Pareto Front Visualization
Basic Pareto Plot
dw$plot()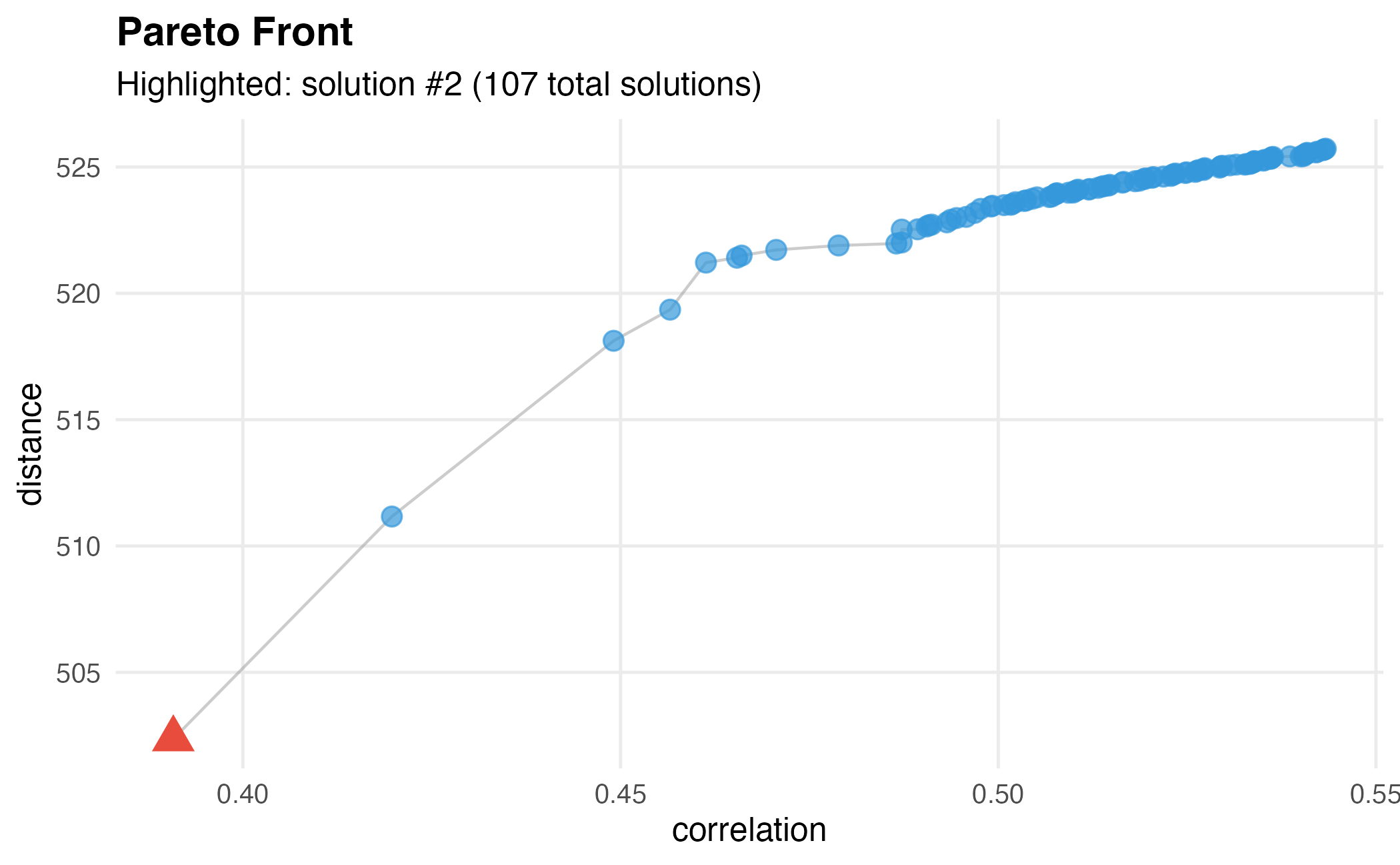
Basic Pareto front visualization showing the trade-off between objectives.
Customized Pareto Plot
# Highlight solution with best distance
dw$plot(
index = c(2, -1), # Objective 2, last rank (highest distance)
point_size = 4,
highlight_size = 7
)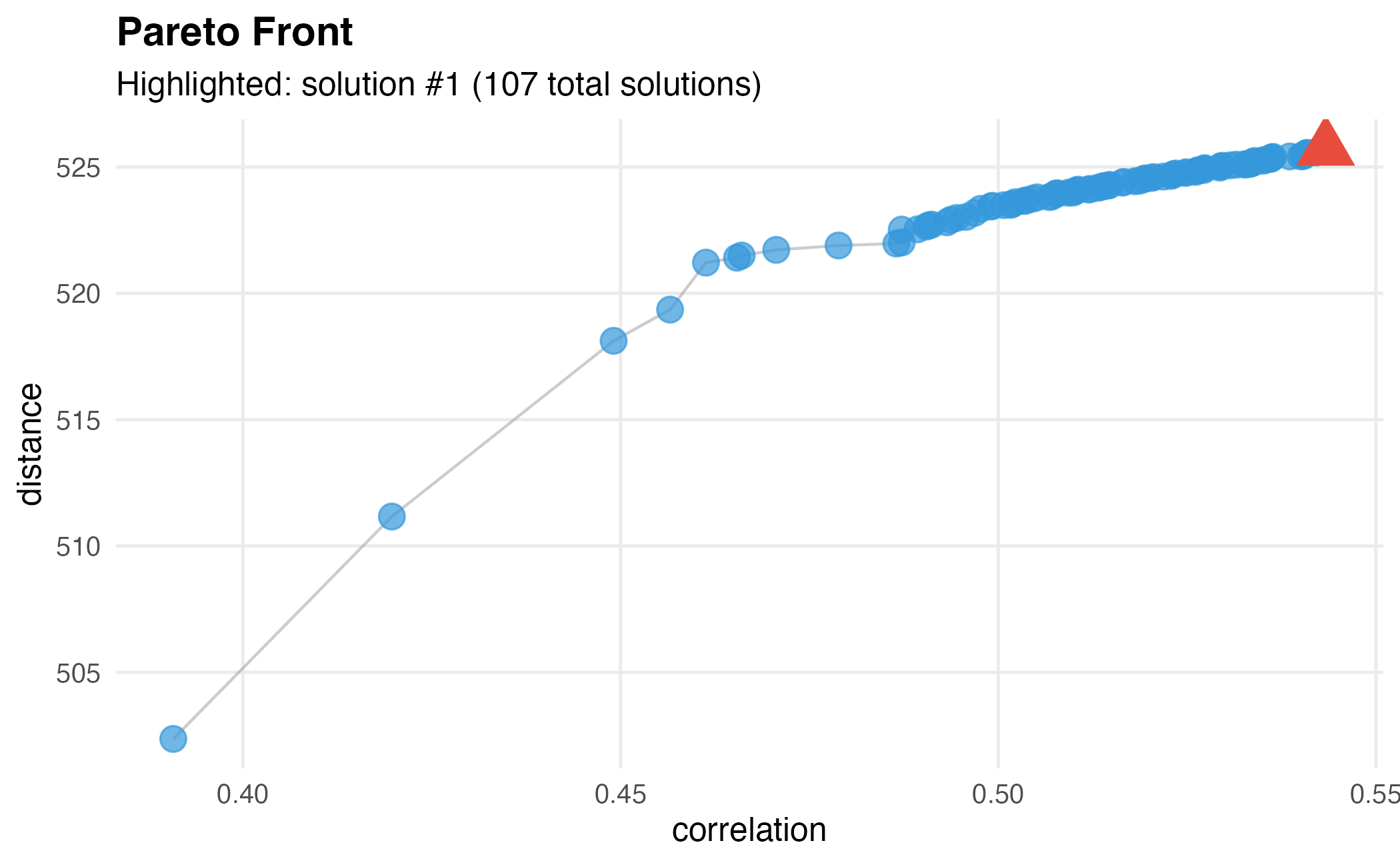
Customized Pareto front with different highlighted solution.
Manual Pareto Plot with ggplot2
# Get fitness data
fitness <- dw$get_fitness()
pareto <- dw$get_pareto()
n_genes_per_solution <- sapply(pareto, sum)
# Create data frame
df <- data.frame(
correlation = fitness$correlation,
distance = fitness$distance,
n_genes = n_genes_per_solution,
solution_id = 1:nrow(fitness)
)
# Custom plot
ggplot(df, aes(x = correlation, y = distance)) +
geom_point(aes(size = n_genes, color = n_genes), alpha = 0.7) +
geom_line(color = "gray50", alpha = 0.5) +
scale_color_viridis_c(option = "plasma") +
scale_size_continuous(range = c(2, 8)) +
labs(
title = "Pareto Front Analysis",
subtitle = paste(nrow(df), "Pareto-optimal solutions"),
x = "Correlation (lower is better)",
y = "Distance (higher is better)",
color = "Number of\nGenes",
size = "Number of\nGenes"
) +
theme_minimal(base_size = 12) +
theme(
plot.title = element_text(face = "bold"),
legend.position = "right"
)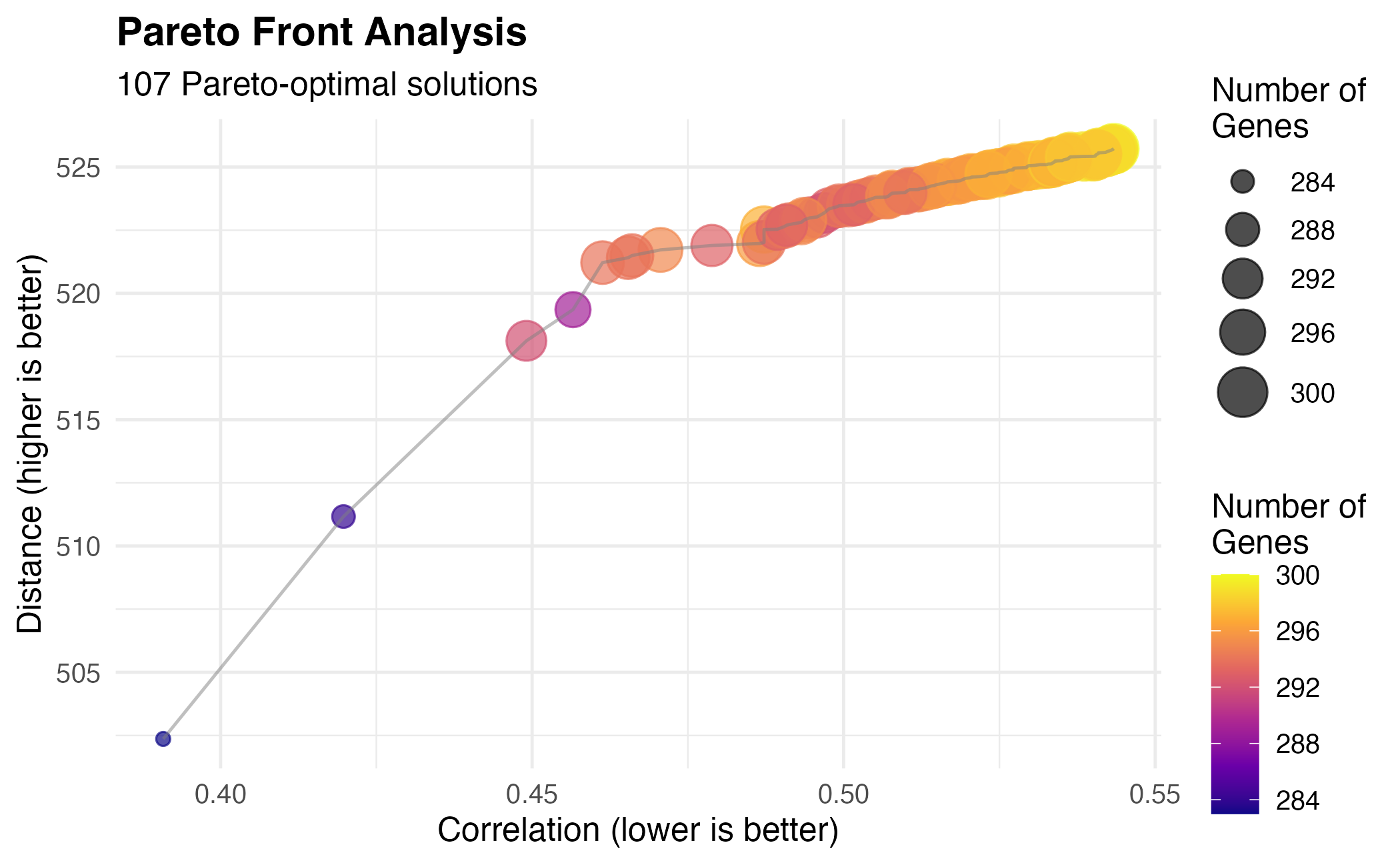
Fully customized Pareto front visualization.
Gene Selection Analysis
Gene Count Distribution
df_genes <- data.frame(n_genes = n_genes_per_solution)
ggplot(df_genes, aes(x = n_genes)) +
geom_histogram(bins = 20, fill = "#3498db", color = "white", alpha = 0.8) +
geom_vline(xintercept = median(n_genes_per_solution),
color = "#e74c3c", linetype = "dashed", linewidth = 1) +
annotate("text", x = median(n_genes_per_solution) + 5, y = Inf,
label = paste("Median:", median(n_genes_per_solution)),
vjust = 2, color = "#e74c3c") +
labs(
title = "Distribution of Selected Gene Counts",
subtitle = "Across all Pareto-optimal solutions",
x = "Number of Selected Genes",
y = "Frequency"
) +
theme_minimal(base_size = 12)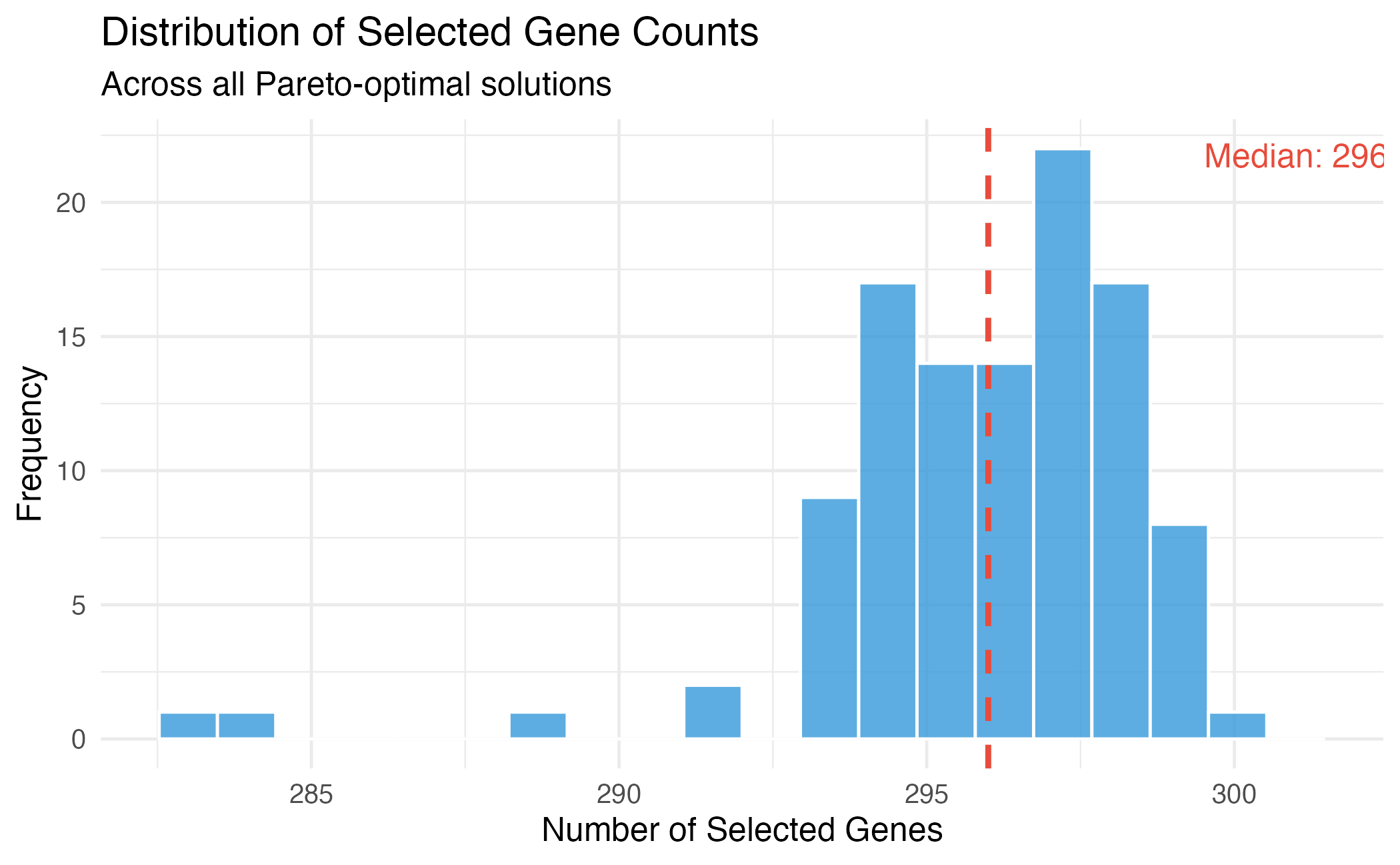
Distribution of selected gene counts across Pareto-optimal solutions.
Fitness vs Gene Count
library(ggplot2)
# Long format for faceting
df_long <- rbind(
data.frame(n_genes = n_genes_per_solution,
value = fitness$correlation,
objective = "Correlation"),
data.frame(n_genes = n_genes_per_solution,
value = fitness$distance,
objective = "Distance")
)
ggplot(df_long, aes(x = n_genes, y = value)) +
geom_point(alpha = 0.6, color = "#3498db") +
geom_smooth(method = "loess", se = TRUE, color = "#e74c3c") +
facet_wrap(~objective, scales = "free_y") +
labs(
title = "Objectives vs Number of Selected Genes",
x = "Number of Selected Genes",
y = "Objective Value"
) +
theme_minimal(base_size = 12)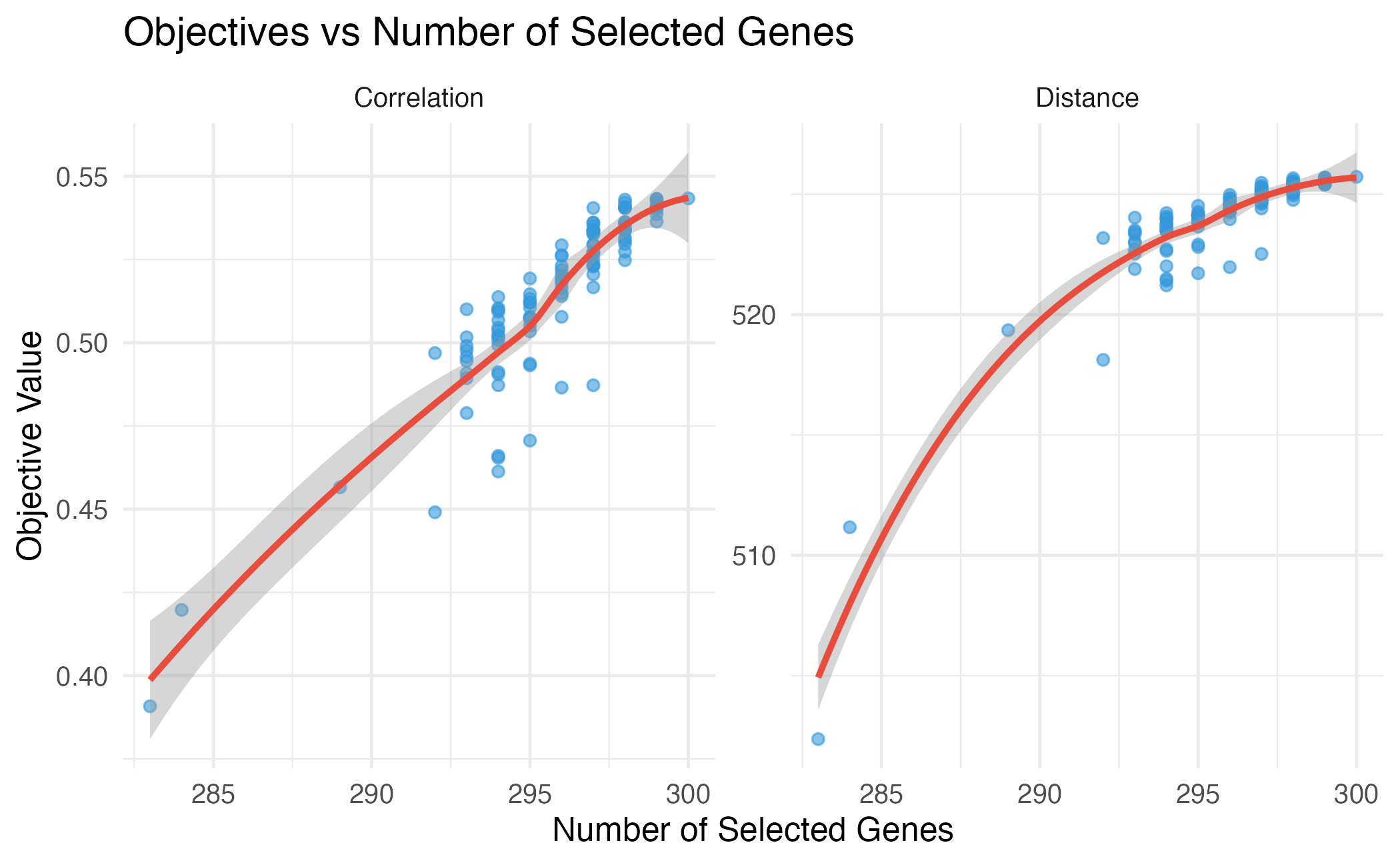
Relationship between number of genes and objective values.
Expression Profile Visualization
Heatmap of Selected Genes
# Select a solution
dw$select(weights = c(-1, 1))
selection <- dw$get_selection()
selected_data <- reference[, selection]
# For visualization, show top 50 most variable genes
gene_vars <- apply(selected_data, 2, var)
top_genes <- names(sort(gene_vars, decreasing = TRUE))[1:min(50, ncol(selected_data))]
plot_data <- selected_data[, top_genes]
# Scale for visualization
plot_data_scaled <- t(scale(t(plot_data)))
# Convert to long format
df_heat <- expand.grid(
CellType = rownames(plot_data_scaled),
Gene = colnames(plot_data_scaled)
)
df_heat$Expression <- as.vector(plot_data_scaled)
ggplot(df_heat, aes(x = Gene, y = CellType, fill = Expression)) +
geom_tile() +
scale_fill_gradient2(low = "#3498db", mid = "white", high = "#e74c3c",
midpoint = 0, limits = c(-3, 3), oob = scales::squish) +
labs(
title = "Expression Heatmap of Selected Genes",
subtitle = paste("Top", length(top_genes), "most variable genes"),
x = "Genes",
y = "Cell Type",
fill = "Scaled\nExpression"
) +
theme_minimal(base_size = 10) +
theme(
axis.text.x = element_blank(),
axis.ticks.x = element_blank()
)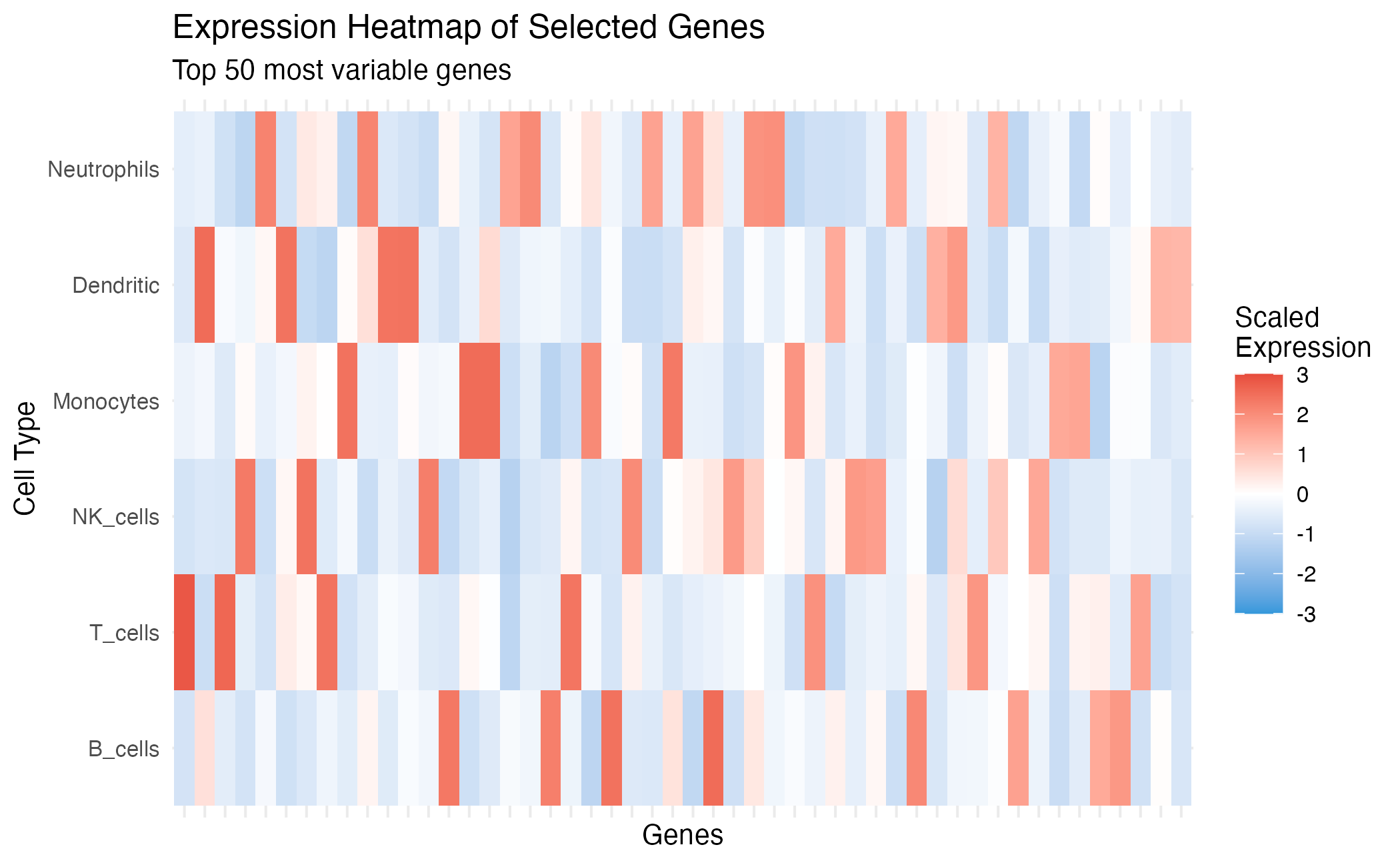
Expression heatmap of selected marker genes.
Cell Type Similarity
# Compute correlation matrix
corr_selected <- cor(t(selected_data))
df_corr <- expand.grid(
CT1 = rownames(corr_selected),
CT2 = colnames(corr_selected)
)
df_corr$Correlation <- as.vector(corr_selected)
ggplot(df_corr, aes(x = CT1, y = CT2, fill = Correlation)) +
geom_tile() +
geom_text(aes(label = round(Correlation, 2)), size = 3) +
scale_fill_gradient2(low = "#3498db", mid = "white", high = "#e74c3c",
midpoint = 0, limits = c(-1, 1)) +
labs(
title = "Cell Type Correlation Matrix",
subtitle = "Based on selected marker genes",
x = "", y = ""
) +
theme_minimal(base_size = 12) +
coord_fixed() +
theme(axis.text.x = element_text(angle = 45, hjust = 1))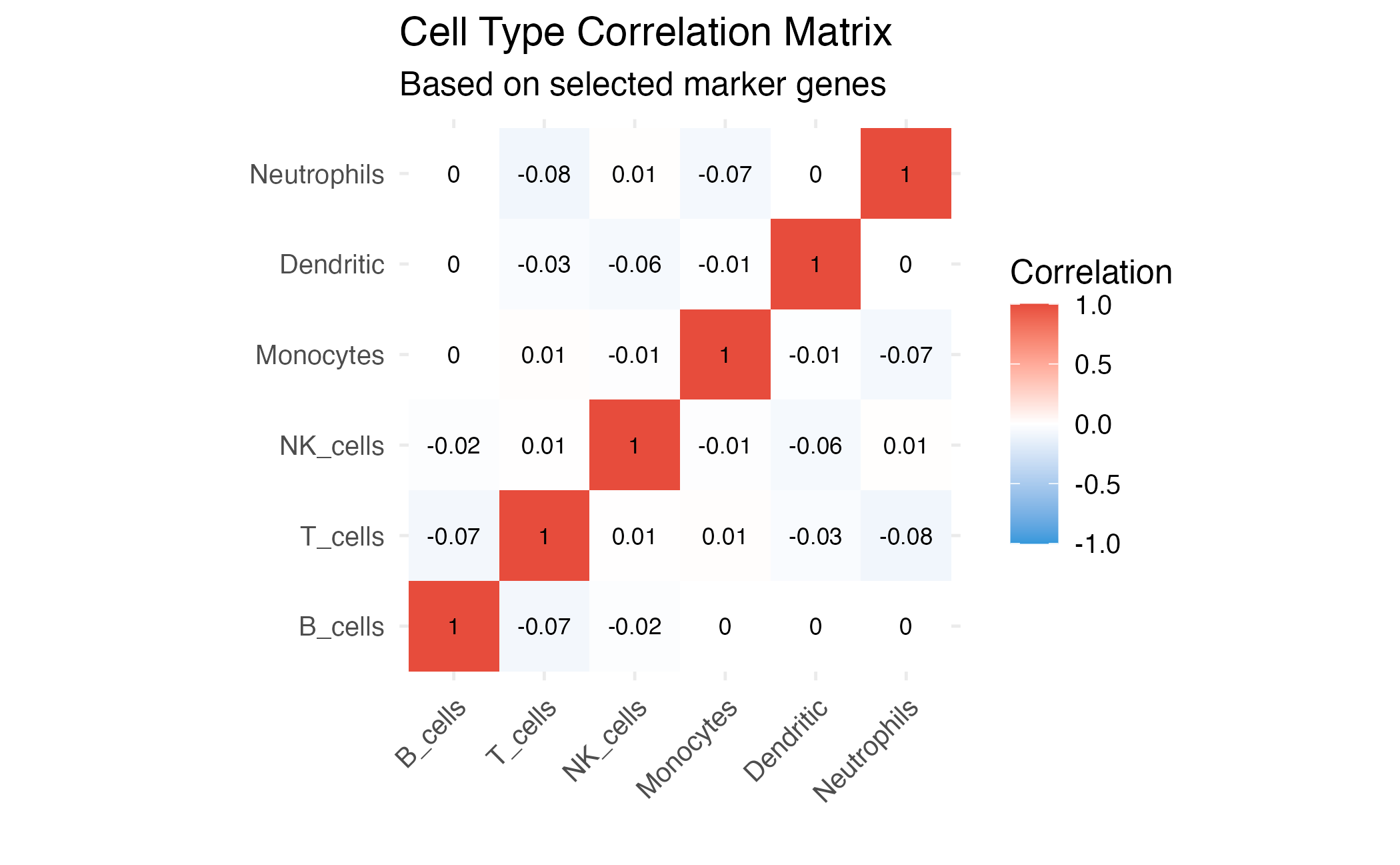
Cell type similarity based on selected genes.
Solution Comparison
Compare Different Selection Methods
# Get multiple solutions
dw$select(weights = c(-1, 1))
sol_weighted <- dw$get_selection()
dw$select(index = 1)
sol_first <- dw$get_selection()
dw$select(index = c(1, 1)) # Best correlation
sol_best_corr <- dw$get_selection()
dw$select(index = c(2, -1)) # Best distance
sol_best_dist <- dw$get_selection()
# Compare
comparison <- data.frame(
Method = c("Weighted", "First", "Best Correlation", "Best Distance"),
N_Genes = c(sum(sol_weighted), sum(sol_first), sum(sol_best_corr), sum(sol_best_dist)),
Correlation = c(
compute_correlation(reference[, sol_weighted]),
compute_correlation(reference[, sol_first]),
compute_correlation(reference[, sol_best_corr]),
compute_correlation(reference[, sol_best_dist])
),
Distance = c(
compute_distance(reference[, sol_weighted]),
compute_distance(reference[, sol_first]),
compute_distance(reference[, sol_best_corr]),
compute_distance(reference[, sol_best_dist])
)
)
knitr::kable(comparison, digits = 2, caption = "Comparison of different selection methods")| Method | N_Genes | Correlation | Distance |
|---|---|---|---|
| Weighted | 283 | 0.39 | 502.36 |
| First | 300 | 0.54 | 525.72 |
| Best Correlation | 283 | 0.39 | 502.36 |
| Best Distance | 300 | 0.54 | 525.72 |
# Plot comparison
df_comp <- data.frame(
Correlation = comparison$Correlation,
Distance = comparison$Distance,
Method = comparison$Method
)
ggplot(df, aes(x = correlation, y = distance)) +
geom_point(color = "gray70", alpha = 0.5, size = 2) +
geom_line(color = "gray70", alpha = 0.5) +
geom_point(data = df_comp, aes(x = Correlation, y = Distance, color = Method),
size = 5) +
scale_color_brewer(palette = "Set1") +
labs(
title = "Solution Comparison on Pareto Front",
x = "Correlation",
y = "Distance"
) +
theme_minimal(base_size = 12) +
theme(legend.position = "bottom")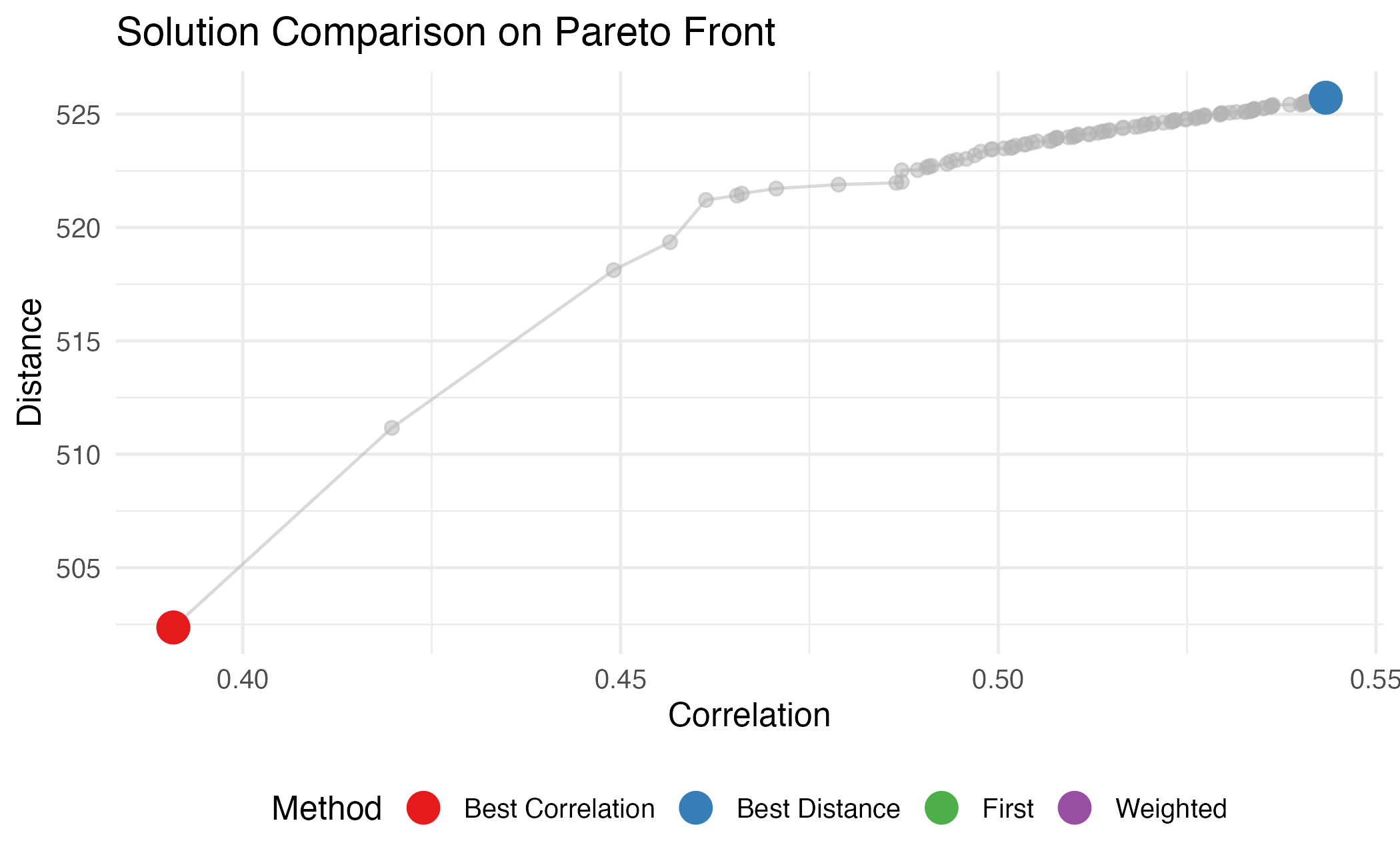
Visual comparison of selection methods on the Pareto front.
Advanced: 3D Pareto Front
For three objectives, we can visualize in 3D:
# Example with 3 objectives (not run)
dw3 <- darwin(reference)
dw3$optimize(
ngen = 50,
objectives = c("correlation", "distance", "condition"),
weights = c(-1, 1, -1),
verbose = FALSE
)
# Would use plotly for 3D visualization
# library(plotly)
# fitness3 <- dw3$get_fitness()
# plot_ly(fitness3, x = ~correlation, y = ~distance, z = ~condition,
# type = "scatter3d", mode = "markers")Summary
Effective visualization of darwin results helps in:
- Understanding trade-offs between objectives
- Comparing different selection strategies
- Validating that selected genes provide good cell type separation
- Communicating results to collaborators
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggplot2_4.0.1 darwin_1.0.0
#>
#> loaded via a namespace (and not attached):
#> [1] sass_0.4.10 future_1.69.0 generics_0.1.4
#> [4] lattice_0.22-7 listenv_0.10.0 digest_0.6.39
#> [7] magrittr_2.0.4 evaluate_1.0.5 grid_4.4.0
#> [10] RColorBrewer_1.1-3 fastmap_1.2.0 jsonlite_2.0.0
#> [13] Matrix_1.7-4 mgcv_1.9-3 viridisLite_0.4.2
#> [16] scales_1.4.0 codetools_0.2-20 textshaping_1.0.4
#> [19] jquerylib_0.1.4 cli_3.6.5 rlang_1.1.7
#> [22] parallelly_1.46.1 future.apply_1.20.1 splines_4.4.0
#> [25] withr_3.0.2 cachem_1.1.0 yaml_2.3.12
#> [28] otel_0.2.0 tools_4.4.0 parallel_4.4.0
#> [31] dplyr_1.1.4 globals_0.18.0 vctrs_0.7.1
#> [34] R6_2.6.1 lifecycle_1.0.5 fs_1.6.6
#> [37] htmlwidgets_1.6.4 ragg_1.5.0 pkgconfig_2.0.3
#> [40] desc_1.4.3 pkgdown_2.1.3 pillar_1.11.1
#> [43] bslib_0.9.0 gtable_0.3.6 glue_1.8.0
#> [46] Rcpp_1.1.1 systemfonts_1.3.1 xfun_0.56
#> [49] tibble_3.3.1 tidyselect_1.2.1 knitr_1.51
#> [52] dichromat_2.0-0.1 farver_2.1.2 nlme_3.1-168
#> [55] htmltools_0.5.9 rmarkdown_2.30 labeling_0.4.3
#> [58] compiler_4.4.0 S7_0.2.1