Bulk RNA-seq Deconvolution
Zaoqu Liu
2026-01-25
Source:vignettes/deconvolution.Rmd
deconvolution.RmdIntroduction
Bulk RNA-seq deconvolution is the computational process of estimating cell type proportions from bulk tissue transcriptomic data. This vignette demonstrates how to use darwin for the complete deconvolution workflow.
The Deconvolution Problem
Mathematical Formulation
The bulk expression of a tissue sample can be modeled as a weighted sum of cell type expression profiles:
where: - : Bulk expression vector (n genes) - : Reference signature matrix (n genes × k cell types) - : Cell type proportions to estimate - : Noise term
Why Gene Selection Matters
The choice of genes significantly impacts deconvolution accuracy:
- Too few genes: Insufficient information for accurate estimation
- Too many genes: Noise and uninformative genes degrade performance
- Correlated genes: Multicollinearity leads to unstable estimates
darwin addresses this by selecting genes that minimize correlation while maximizing cell type separability.
Complete Workflow
Step 2: Create Reference Matrix
In practice, this would come from single-cell RNA-seq data.
# Simulate reference data: 8 cell types × 500 genes
n_celltypes <- 8
n_genes <- 500
cell_types <- c("B_cells", "CD4_T", "CD8_T", "NK", "Monocytes",
"Dendritic", "Macrophages", "Neutrophils")
reference <- matrix(
abs(rnorm(n_celltypes * n_genes, mean = 3, sd = 1.5)),
nrow = n_celltypes,
ncol = n_genes
)
rownames(reference) <- cell_types
colnames(reference) <- paste0("Gene", 1:n_genes)
# Add cell-type specific marker expression
marker_strength <- c(5, 4, 4, 4.5, 3.5, 4, 3.5, 3)
for (i in 1:n_celltypes) {
markers <- ((i - 1) * 20 + 1):(i * 20)
reference[i, markers] <- reference[i, markers] + marker_strength[i]
}
cat("Reference matrix dimensions:", dim(reference), "\n")
#> Reference matrix dimensions: 8 500Step 3: Create Simulated Bulk Data
We create bulk samples with known cell type proportions for validation.
# True proportions for 10 samples
n_samples <- 10
true_proportions <- matrix(0, nrow = n_samples, ncol = n_celltypes)
colnames(true_proportions) <- cell_types
rownames(true_proportions) <- paste0("Sample", 1:n_samples)
# Generate random proportions that sum to 1
for (i in 1:n_samples) {
props <- runif(n_celltypes)
true_proportions[i, ] <- props / sum(props)
}
# Generate bulk expression: bulk = proportions %*% reference + noise
bulk <- true_proportions %*% reference
bulk <- bulk + matrix(rnorm(n_samples * n_genes, sd = 0.5), nrow = n_samples)
bulk[bulk < 0] <- 0
cat("Bulk matrix dimensions:", dim(bulk), "\n")
#> Bulk matrix dimensions: 10 500
cat("\nTrue proportions (first 3 samples):\n")
#>
#> True proportions (first 3 samples):
print(round(true_proportions[1:3, ], 3))
#> B_cells CD4_T CD8_T NK Monocytes Dendritic Macrophages Neutrophils
#> Sample1 0.122 0.090 0.057 0.053 0.102 0.199 0.178 0.199
#> Sample2 0.007 0.022 0.045 0.274 0.175 0.165 0.039 0.274
#> Sample3 0.228 0.039 0.042 0.149 0.053 0.112 0.112 0.265Step 4: Optimize Gene Selection
# Initialize darwin
dw <- darwin(reference)
# Run multi-objective optimization
dw$optimize(
ngen = 100,
pop_size = 80,
objectives = c("correlation", "distance"),
weights = c(-1, 1),
verbose = FALSE,
parallel = FALSE
)
# Visualize Pareto front
dw$plot()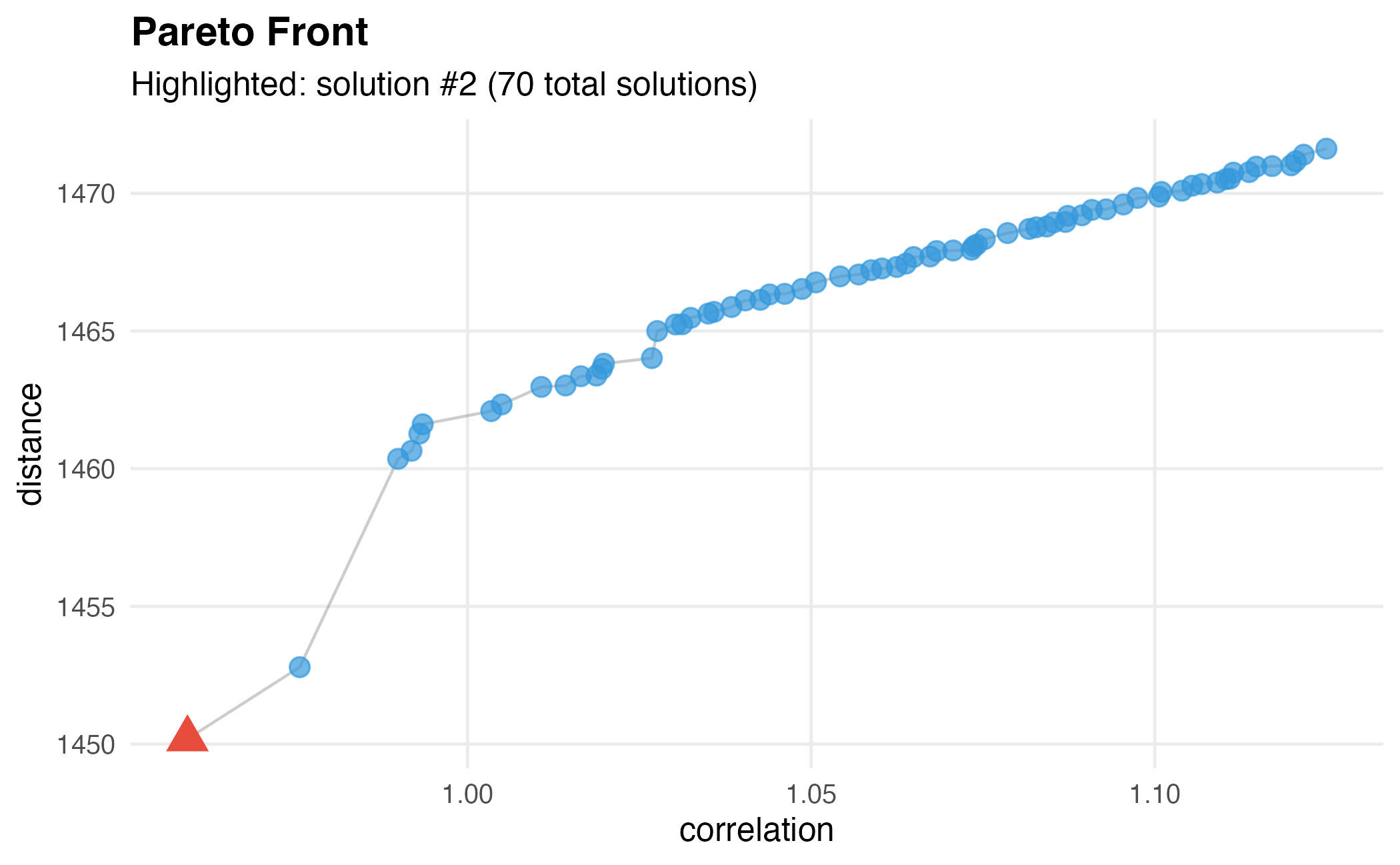
Step 5: Select Optimal Genes
# Select solution balancing both objectives
dw$select(weights = c(-1, 1))
# Check selection statistics
genes <- dw$get_genes()
selection <- dw$get_selection()
cat("Selected", length(genes), "genes\n")
#> Selected 489 genes
cat("Correlation score:", round(compute_correlation(reference[, selection]), 3), "\n")
#> Correlation score: 0.959
cat("Distance score:", round(compute_distance(reference[, selection]), 3), "\n")
#> Distance score: 1450.187Step 6: Perform Deconvolution
darwin supports three deconvolution methods:
Non-Negative Least Squares (NNLS)
result_nnls <- dw$deconvolve(bulk, method = "nnls")
cat("Estimated proportions (NNLS):\n")
#> Estimated proportions (NNLS):
print(round(result_nnls$proportions[1:3, ], 3))
#> B_cells CD4_T CD8_T NK Monocytes Dendritic Macrophages Neutrophils
#> Sample1 0.103 0.091 0.065 0.073 0.118 0.189 0.164 0.196
#> Sample2 0.001 0.037 0.055 0.270 0.189 0.163 0.026 0.258
#> Sample3 0.211 0.038 0.056 0.160 0.056 0.114 0.116 0.249Linear Regression
result_linear <- dw$deconvolve(bulk, method = "linear")
cat("Estimated proportions (Linear):\n")
#> Estimated proportions (Linear):
print(round(result_linear$proportions[1:3, ], 3))
#> B_cells CD4_T CD8_T NK Monocytes Dendritic Macrophages Neutrophils
#> Sample1 0.103 0.091 0.065 0.073 0.118 0.189 0.164 0.196
#> Sample2 0.001 0.037 0.055 0.270 0.189 0.163 0.026 0.258
#> Sample3 0.211 0.038 0.056 0.160 0.056 0.114 0.116 0.249Step 7: Evaluate Accuracy
# Compare estimated to true proportions
estimated <- result_nnls$proportions
# Calculate correlation per cell type
correlations <- sapply(1:n_celltypes, function(i) {
cor(true_proportions[, i], estimated[, i])
})
names(correlations) <- cell_types
# Calculate RMSE per cell type
rmse <- sapply(1:n_celltypes, function(i) {
sqrt(mean((true_proportions[, i] - estimated[, i])^2))
})
names(rmse) <- cell_types
# Summary table
accuracy <- data.frame(
CellType = cell_types,
Correlation = round(correlations, 3),
RMSE = round(rmse, 4)
)
knitr::kable(accuracy, caption = "Deconvolution accuracy by cell type")| CellType | Correlation | RMSE | |
|---|---|---|---|
| B_cells | B_cells | 0.984 | 0.0152 |
| CD4_T | CD4_T | 0.994 | 0.0158 |
| CD8_T | CD8_T | 0.996 | 0.0094 |
| NK | NK | 0.986 | 0.0142 |
| Monocytes | Monocytes | 0.994 | 0.0108 |
| Dendritic | Dendritic | 0.971 | 0.0136 |
| Macrophages | Macrophages | 0.987 | 0.0135 |
| Neutrophils | Neutrophils | 0.990 | 0.0100 |
# Create comparison data frame
df_compare <- data.frame(
True = as.vector(true_proportions),
Estimated = as.vector(estimated),
CellType = rep(cell_types, each = n_samples),
Sample = rep(rownames(true_proportions), n_celltypes)
)
# Overall correlation
overall_cor <- cor(df_compare$True, df_compare$Estimated)
ggplot(df_compare, aes(x = True, y = Estimated, color = CellType)) +
geom_point(size = 2, alpha = 0.7) +
geom_abline(intercept = 0, slope = 1, linetype = "dashed", color = "gray40") +
scale_color_brewer(palette = "Set2") +
labs(
title = "True vs Estimated Cell Type Proportions",
subtitle = paste("Overall Pearson r =", round(overall_cor, 3)),
x = "True Proportion",
y = "Estimated Proportion",
color = "Cell Type"
) +
theme_minimal(base_size = 12) +
coord_fixed(xlim = c(0, 0.4), ylim = c(0, 0.4))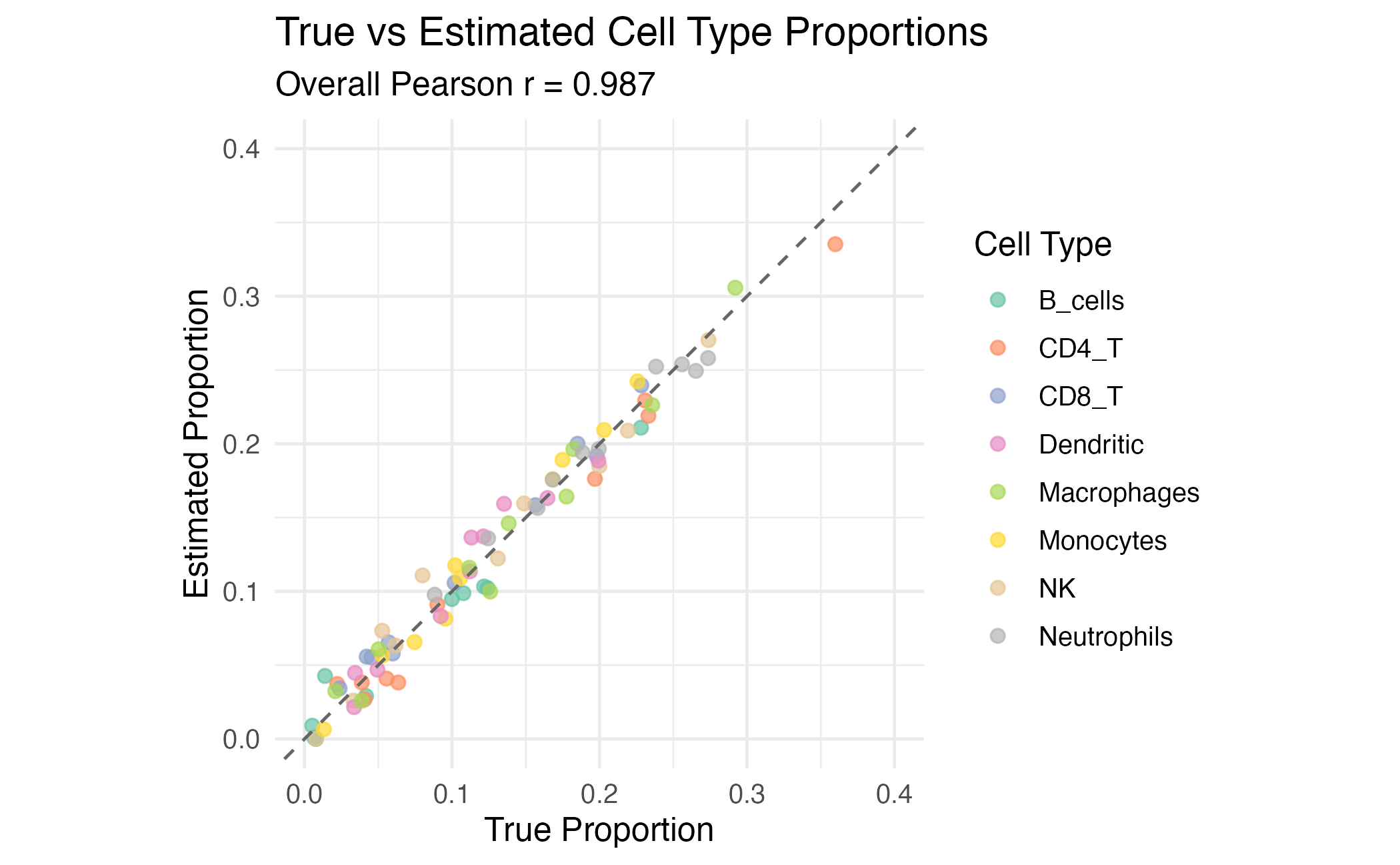
Scatter plot of true vs estimated proportions for each cell type.
ggplot(df_compare, aes(x = True, y = Estimated)) +
geom_point(color = "#3498db", alpha = 0.7) +
geom_abline(intercept = 0, slope = 1, linetype = "dashed", color = "#e74c3c") +
facet_wrap(~CellType, scales = "free") +
labs(
title = "Deconvolution Accuracy by Cell Type",
x = "True Proportion",
y = "Estimated Proportion"
) +
theme_minimal(base_size = 10)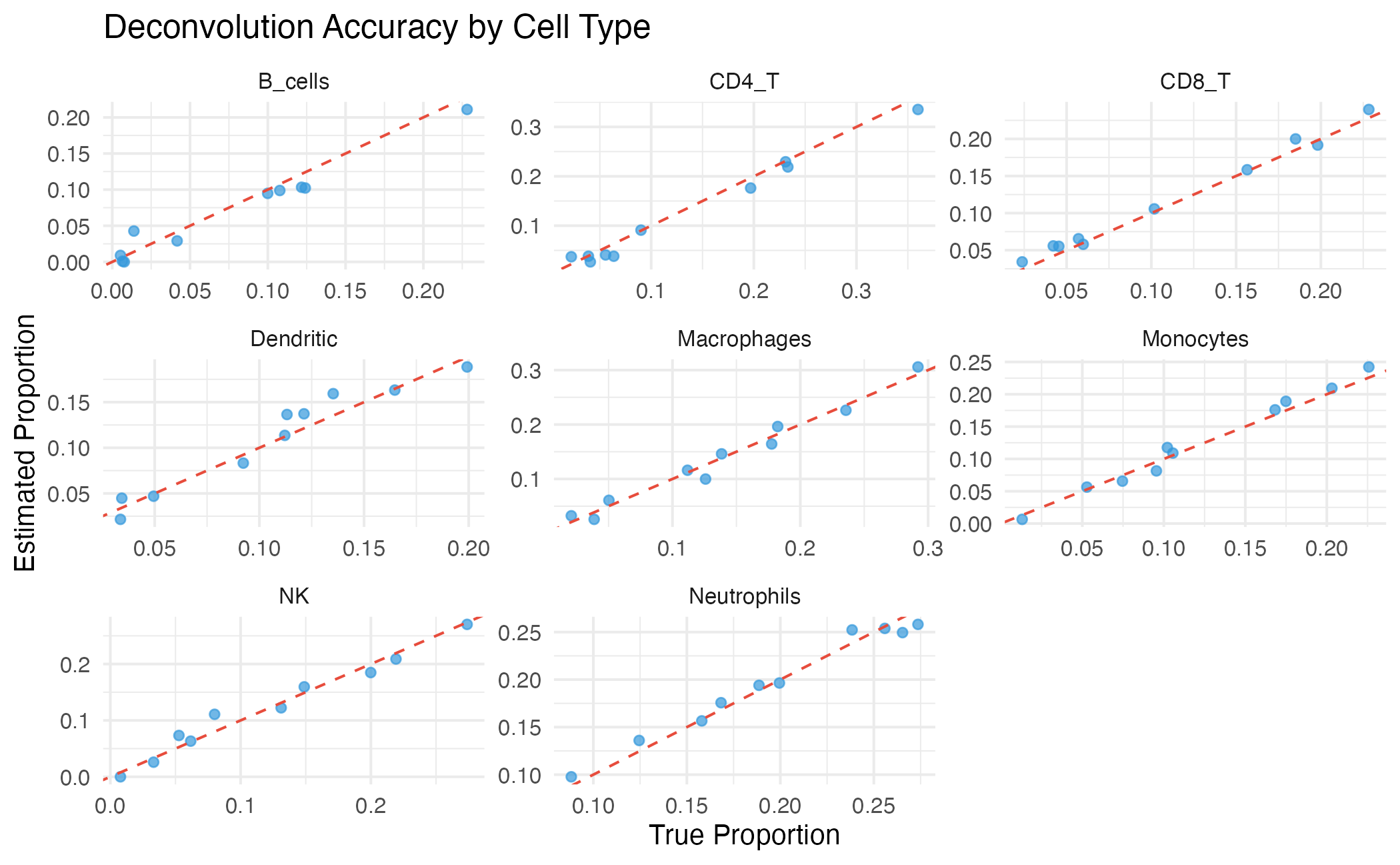
Per-cell-type comparison of true vs estimated proportions.
Comparing Gene Selection Strategies
# Strategy 1: All genes
all_genes_cor <- sapply(1:n_celltypes, function(i) {
# Deconvolve with all genes (simplified)
X <- t(reference)
y <- t(bulk)
coef_mat <- solve(t(X) %*% X) %*% t(X) %*% y
estimated_all <- t(coef_mat)
estimated_all <- estimated_all / rowSums(abs(estimated_all))
cor(true_proportions[, i], estimated_all[, i])
})
# Strategy 2: darwin selected genes
darwin_cor <- correlations
# Strategy 3: Random genes (same number)
n_selected <- sum(selection)
set.seed(123)
random_selection <- sample(1:n_genes, n_selected)
X_random <- t(reference[, random_selection])
y_random <- t(bulk[, random_selection])
coef_random <- solve(t(X_random) %*% X_random) %*% t(X_random) %*% y_random
estimated_random <- t(coef_random)
estimated_random <- estimated_random / rowSums(abs(estimated_random))
random_cor <- sapply(1:n_celltypes, function(i) {
cor(true_proportions[, i], estimated_random[, i])
})
# Compare
comparison <- data.frame(
CellType = rep(cell_types, 3),
Correlation = c(all_genes_cor, darwin_cor, random_cor),
Strategy = rep(c("All Genes", "darwin", "Random"), each = n_celltypes)
)
ggplot(comparison, aes(x = CellType, y = Correlation, fill = Strategy)) +
geom_bar(stat = "identity", position = "dodge") +
scale_fill_manual(values = c("#95a5a6", "#3498db", "#e74c3c")) +
labs(
title = "Deconvolution Accuracy by Gene Selection Strategy",
x = "Cell Type",
y = "Correlation (True vs Estimated)"
) +
theme_minimal(base_size = 12) +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
coord_cartesian(ylim = c(0, 1))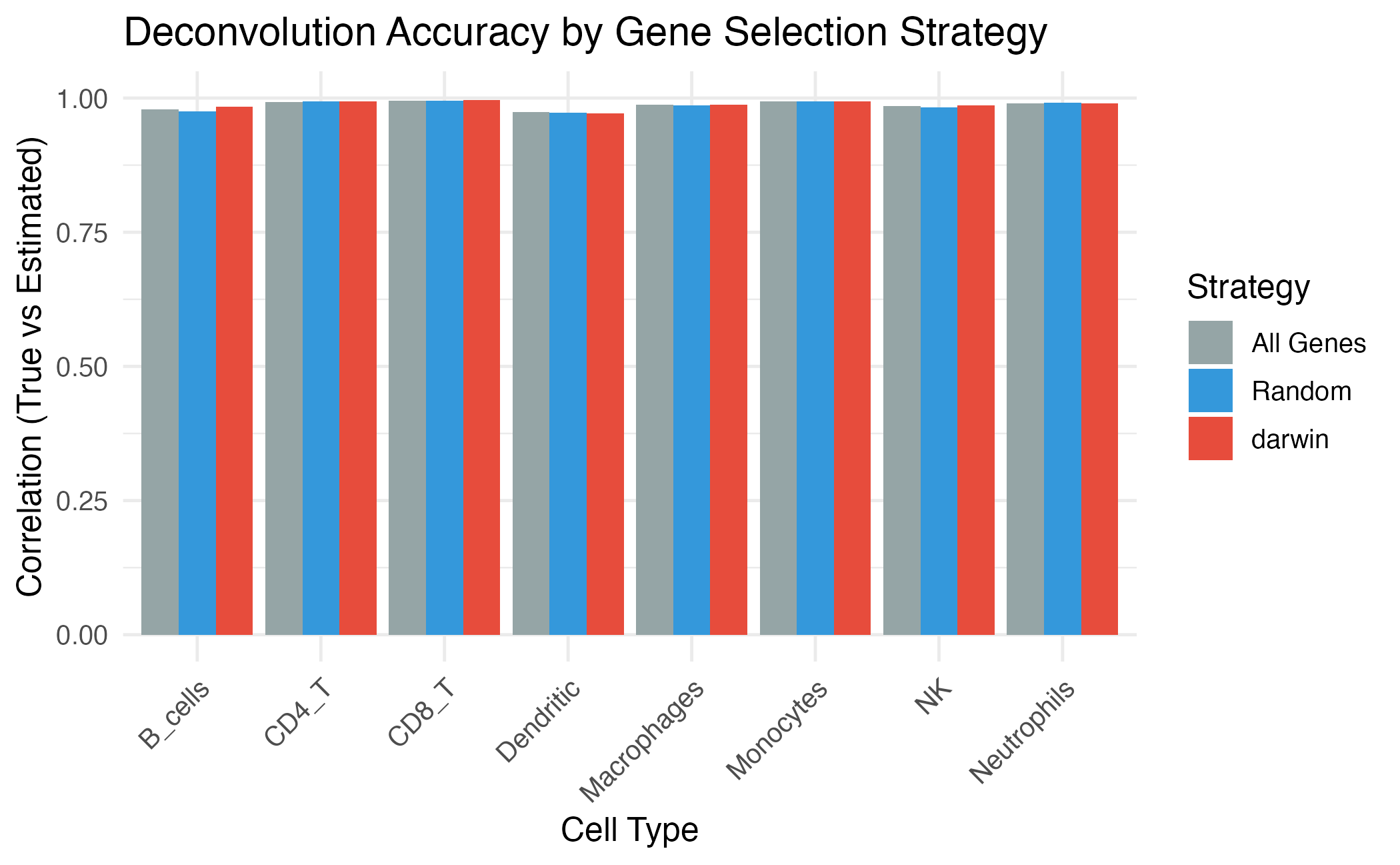
Impact of gene selection on deconvolution accuracy.
Best Practices
1. Reference Matrix Quality
- Use high-quality single-cell reference data
- Ensure all relevant cell types are represented
- Consider batch effects between reference and bulk data
2. Gene Selection
- Run optimization with sufficient generations (≥100)
- Compare multiple solutions from the Pareto front
- Validate with known mixtures when possible
Session Info
sessionInfo()
#> R version 4.4.0 (2024-04-24)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS 15.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] C
#>
#> time zone: Asia/Shanghai
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggplot2_4.0.1 darwin_1.0.0
#>
#> loaded via a namespace (and not attached):
#> [1] sass_0.4.10 future_1.69.0 generics_0.1.4
#> [4] lattice_0.22-7 listenv_0.10.0 digest_0.6.39
#> [7] magrittr_2.0.4 evaluate_1.0.5 grid_4.4.0
#> [10] RColorBrewer_1.1-3 fastmap_1.2.0 jsonlite_2.0.0
#> [13] Matrix_1.7-4 scales_1.4.0 codetools_0.2-20
#> [16] textshaping_1.0.4 jquerylib_0.1.4 cli_3.6.5
#> [19] rlang_1.1.7 parallelly_1.46.1 future.apply_1.20.1
#> [22] withr_3.0.2 cachem_1.1.0 yaml_2.3.12
#> [25] otel_0.2.0 tools_4.4.0 parallel_4.4.0
#> [28] dplyr_1.1.4 globals_0.18.0 vctrs_0.7.1
#> [31] R6_2.6.1 lifecycle_1.0.5 fs_1.6.6
#> [34] htmlwidgets_1.6.4 ragg_1.5.0 pkgconfig_2.0.3
#> [37] desc_1.4.3 pkgdown_2.1.3 pillar_1.11.1
#> [40] bslib_0.9.0 gtable_0.3.6 glue_1.8.0
#> [43] Rcpp_1.1.1 systemfonts_1.3.1 xfun_0.56
#> [46] tibble_3.3.1 tidyselect_1.2.1 knitr_1.51
#> [49] dichromat_2.0-0.1 farver_2.1.2 htmltools_0.5.9
#> [52] rmarkdown_2.30 labeling_0.4.3 compiler_4.4.0
#> [55] S7_0.2.1 nnls_1.6