Introduction
ggforge is a ggplot2 extension that provides 77 high-level plotting functions with publication-oriented defaults. It is inspired by and modified from plotthis by Panwen Wang, with enhancements for broader scientific coverage and a unified API.
Key Features
- 77 plotting functions across 13 scientific categories
- Unified API:
data,x,y,group_by,split_by,palettework consistently - Intelligent variable type detection and automatic styling
- 200+ color palettes (RColorBrewer, ggsci, viridis, Tableau, and more)
- Multi-panel layouts via
split_by+combinepowered by patchwork - Publication-oriented themes and typography
Installation
install.packages("ggforge", repos = "https://zaoqu-liu.r-universe.dev")Core Concepts
Unified API
All ggforge functions follow a consistent pattern:
PlotFunction(
data, # Data frame or matrix
x, y, # Primary aesthetics
group_by = NULL, # Grouping variable (discrete colors)
split_by = NULL, # Split into multiple panels
palette = "Paired", # Color palette name
theme = "theme_ggforge", # Theme function
... # Function-specific parameters
)Intelligent Type Detection
ggforge detects variable types and applies appropriate styling:
- Continuous → gradient color scales
- Discrete → categorical color palettes
- Temporal → time axis formatting
Split and Combine
Create multi-panel figures with split_by:
BoxPlot(..., split_by = "group", combine = TRUE, nrow = 1)Category 1: Statistics
Scatter Plot
scatter_data <- data.frame(
gene_A = rnorm(100, 8, 2), gene_B = rnorm(100, 8, 2),
tissue = sample(c("Normal", "Tumor"), 100, replace = TRUE)
)
scatter_data$gene_B <- scatter_data$gene_B + 0.6 * scatter_data$gene_A
ScatterPlot(scatter_data, x = "gene_A", y = "gene_B",
group_by = "tissue", palette = "npg",
add_smooth = TRUE, add_stat = TRUE,
title = "Gene Expression Correlation")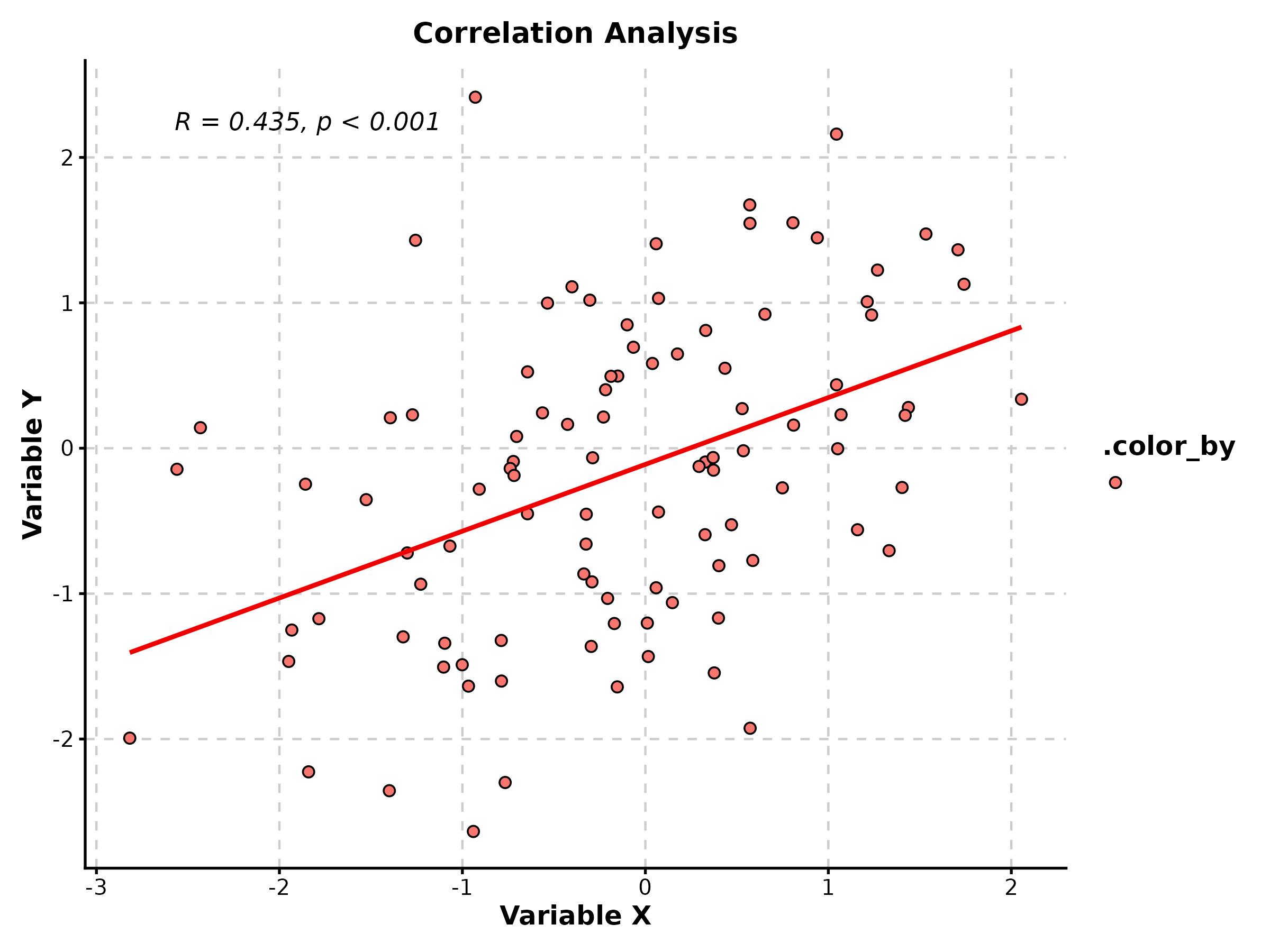
Box Plot
treat_data <- data.frame(
group = rep(c("Control", "Low", "High"), each = 40),
response = c(rnorm(40, 10, 2), rnorm(40, 12, 2.5), rnorm(40, 15, 2))
)
BoxPlot(treat_data, x = "group", y = "response", palette = "lancet",
add_point = TRUE, pt_alpha = 0.3,
comparisons = list(c("Control", "High")),
title = "Treatment Response", ylab = "Expression Level")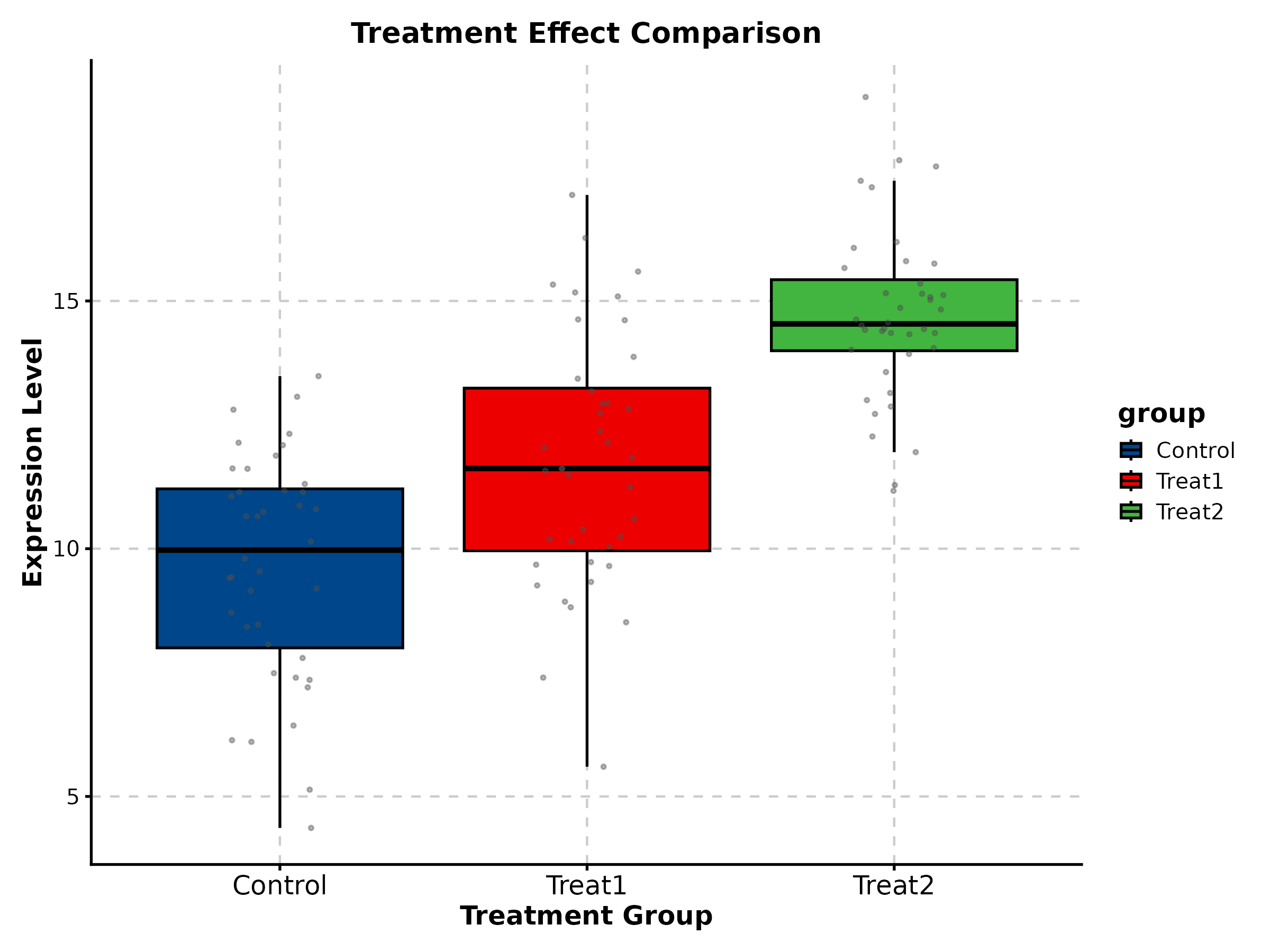
Violin Plot
ViolinPlot(treat_data, x = "group", y = "response", palette = "npg",
add_box = TRUE, add_point = TRUE, pt_alpha = 0.3,
title = "Distribution Comparison")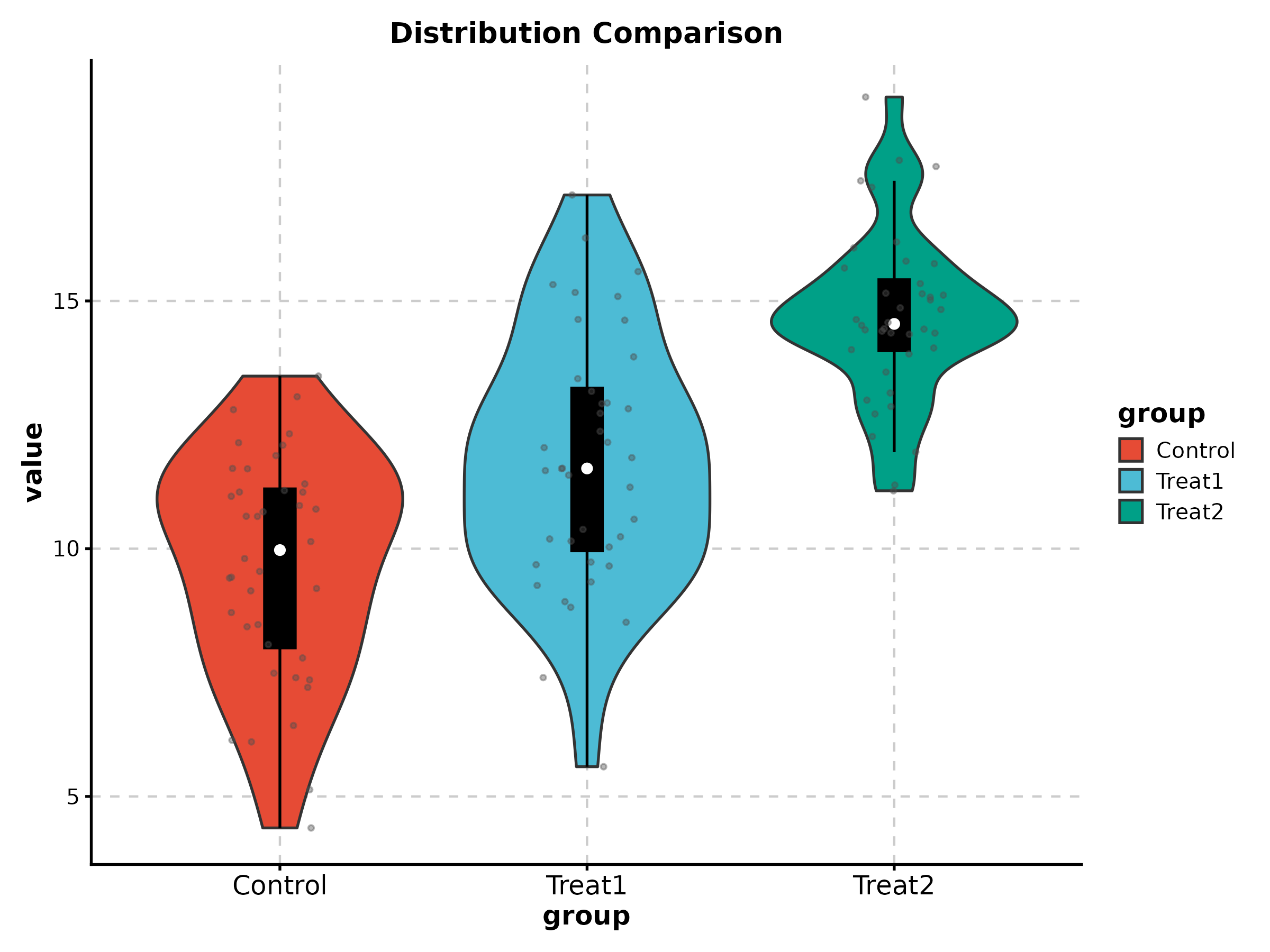
Density Plot
DensityPlot(treat_data, x = "response", group_by = "group",
palette = "npg", add_rug = TRUE,
title = "Response Distribution")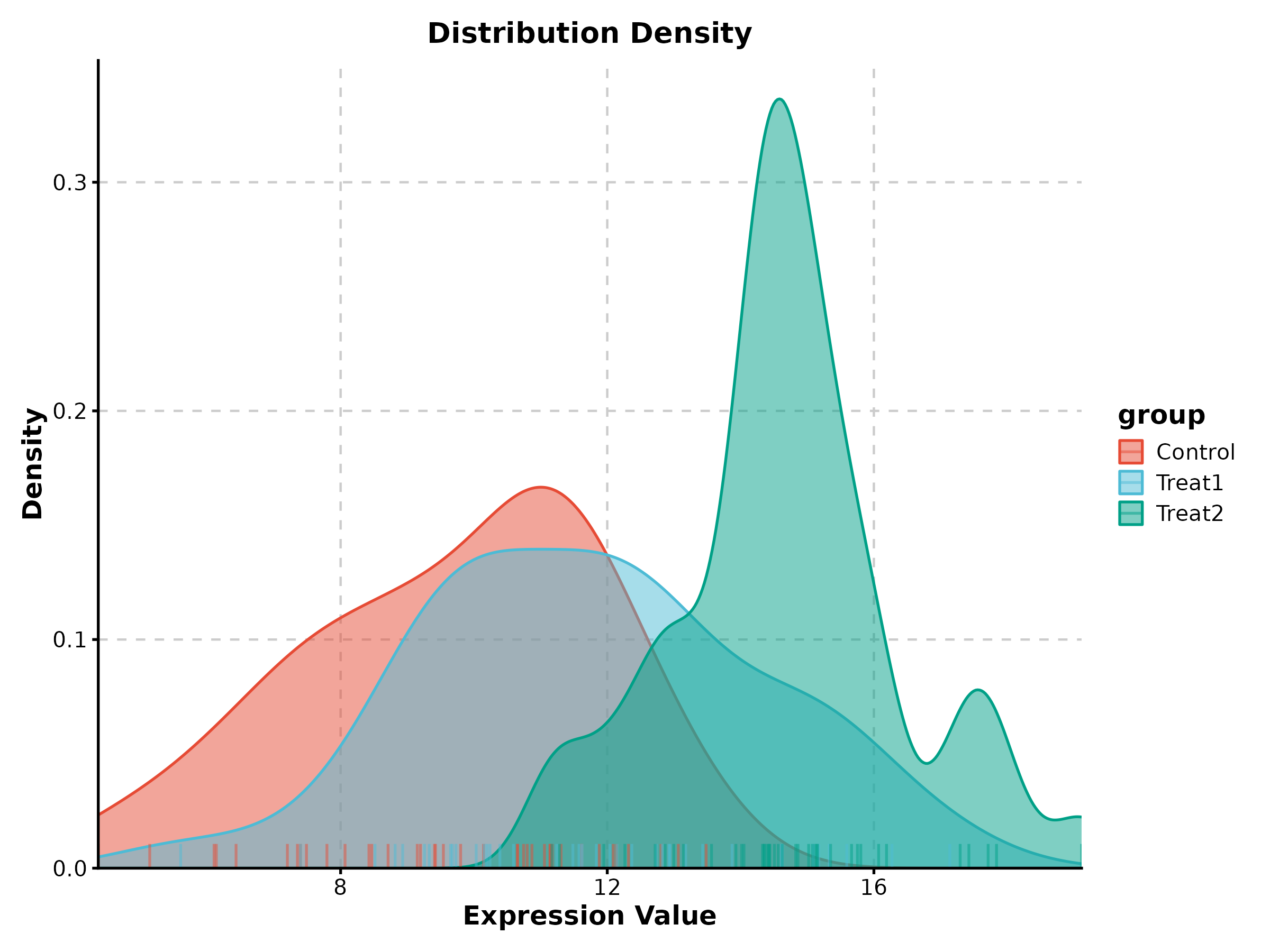
Bar Plot
BarPlot(treat_data, x = "group", y = "response", palette = "Set2",
add_errorbar = TRUE, errorbar_type = "se",
title = "Mean with Standard Error")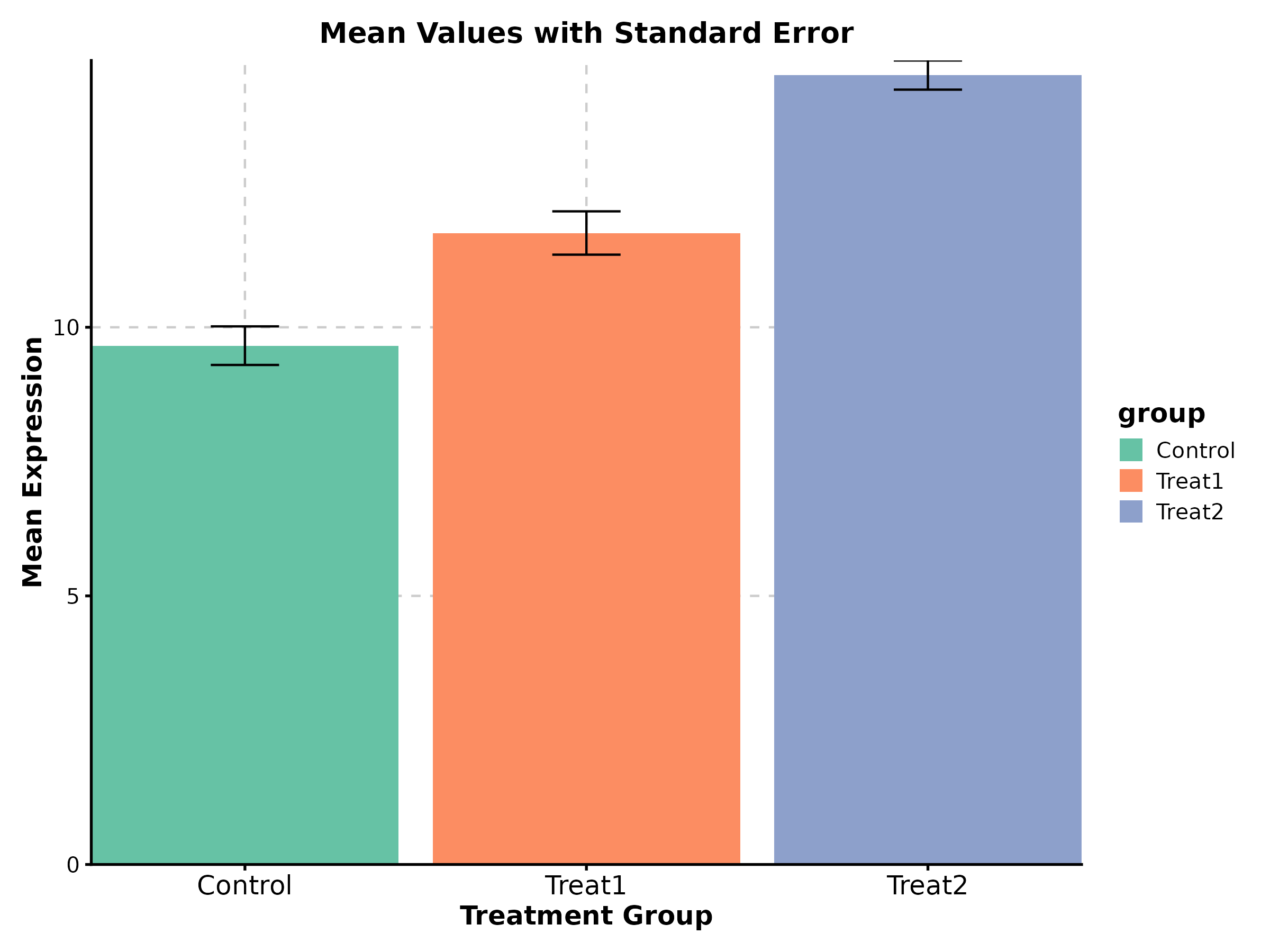
Line Plot
time_data <- data.frame(
day = rep(c(0, 7, 14, 21, 28), 2),
size = c(100, 150, 250, 400, 650, 100, 120, 140, 160, 190),
arm = rep(c("Vehicle", "Treatment"), each = 5)
)
LinePlot(time_data, x = "day", y = "size", group_by = "arm",
palette = "nejm", title = "Tumor Growth",
xlab = "Day", ylab = "Volume (mm³)")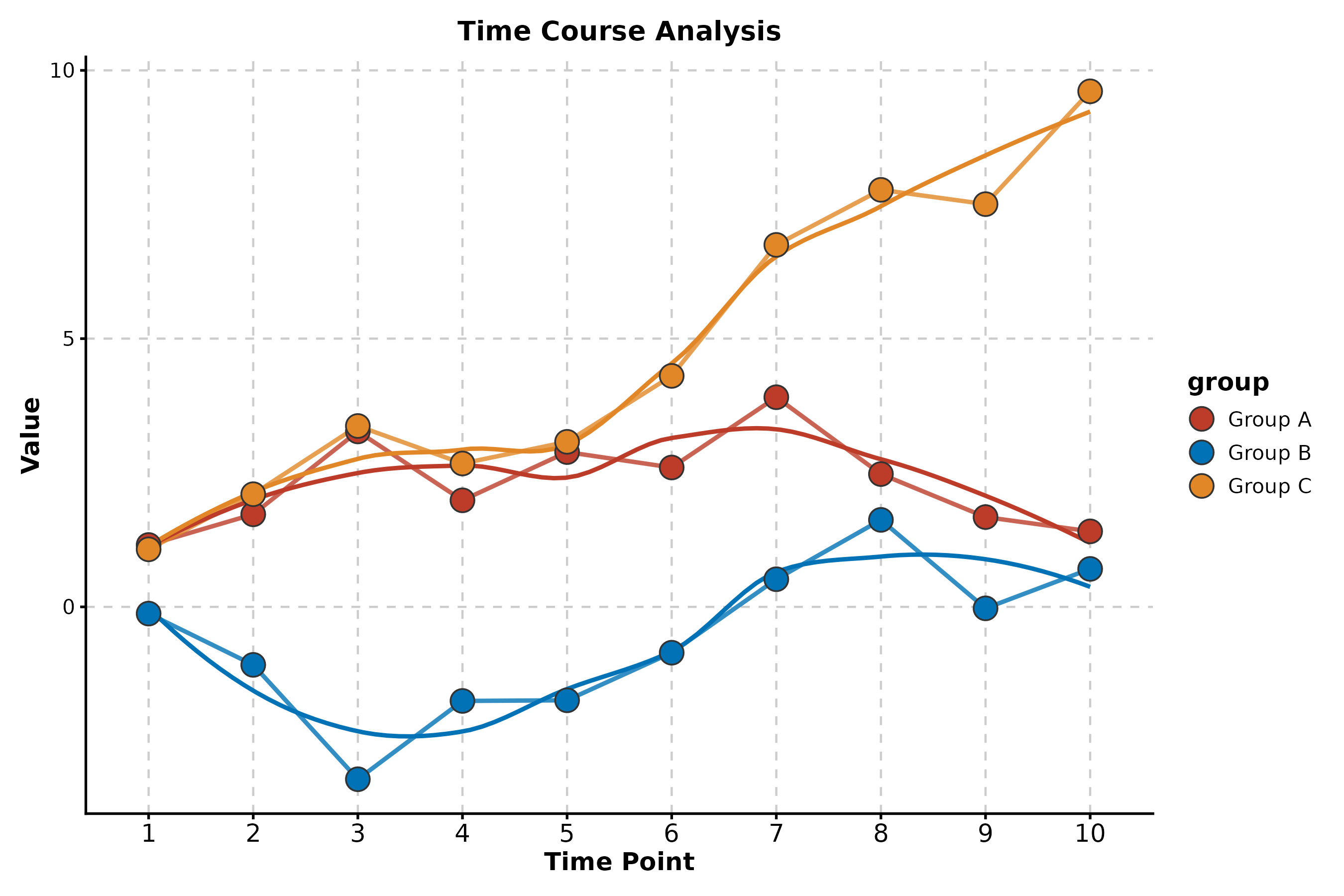
Category 2: Enrichment & Pathway
Enrichment Network
data("enrich_multidb_example")
EnrichNetwork(enrich_multidb_example, top_term = 15,
layout = "fr", palette = "Set3",
title = "Pathway Network")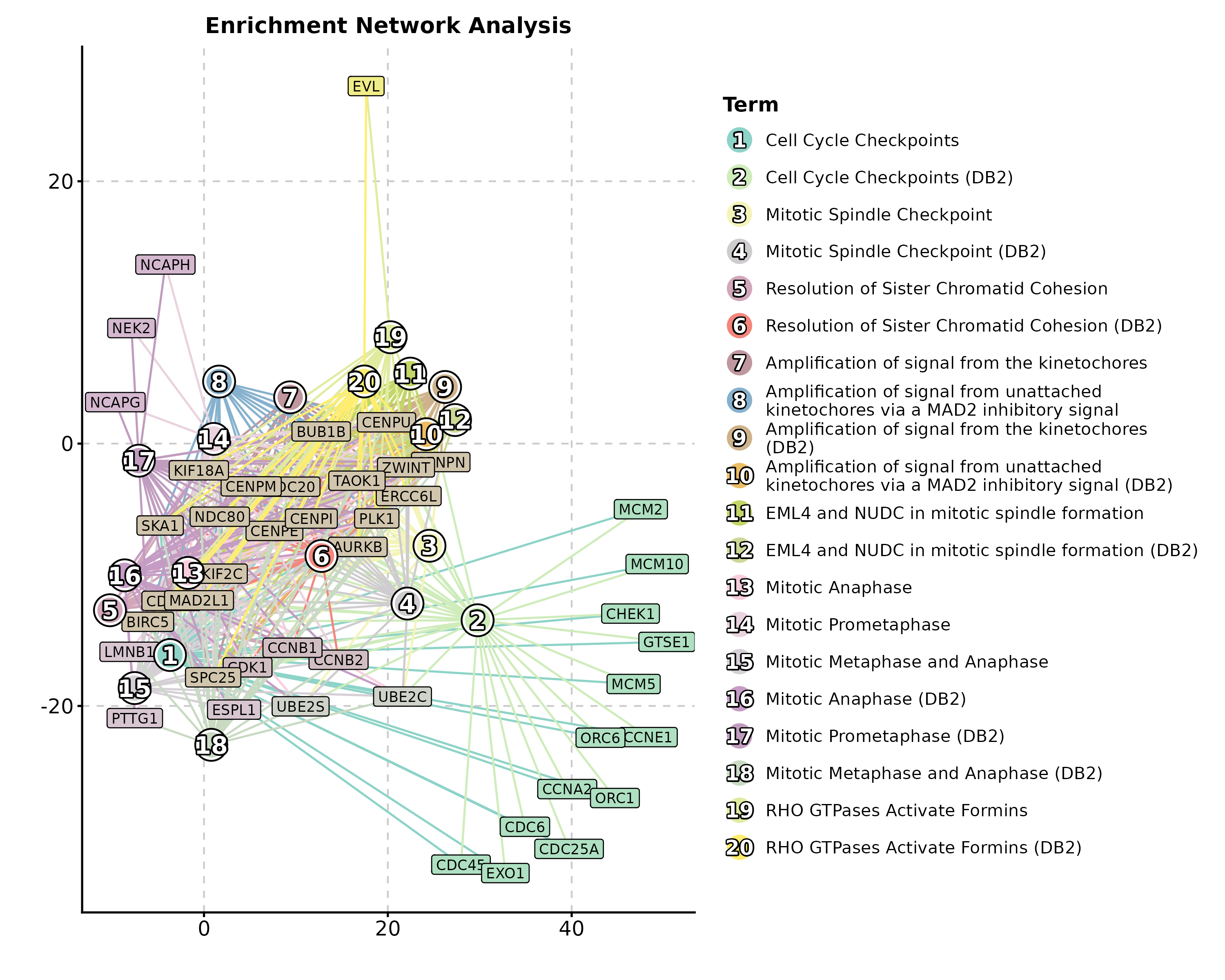
Enrichment Map
data("enrich_example")
EnrichMap(enrich_example, top_term = 20,
layout = "fr", palette = "Spectral",
title = "GO Enrichment Map")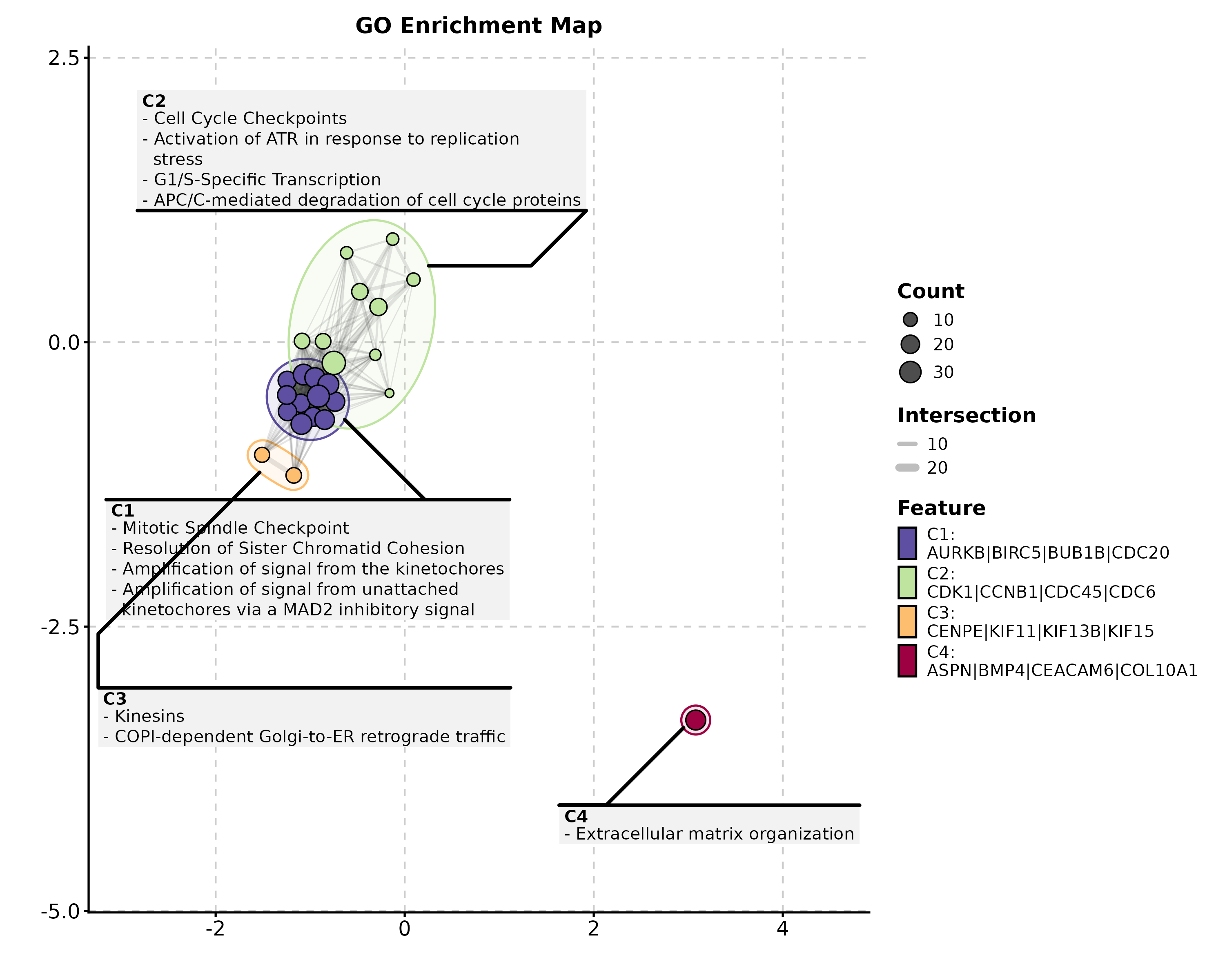
GSEA Summary
data("gsea_example")
GSEASummaryPlot(gsea_example, top_term = 15, palette = "RdBu",
title = "GSEA Results")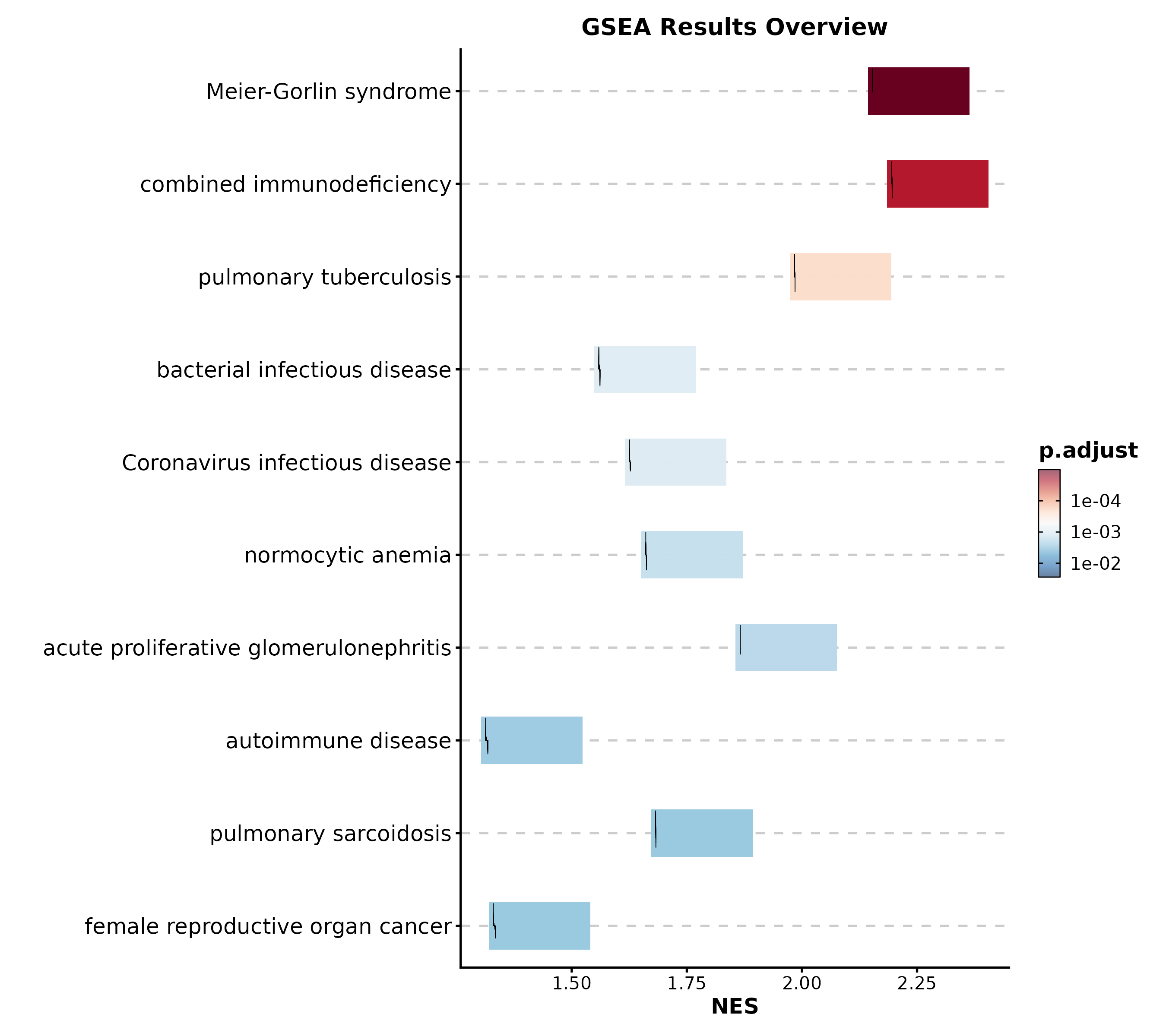
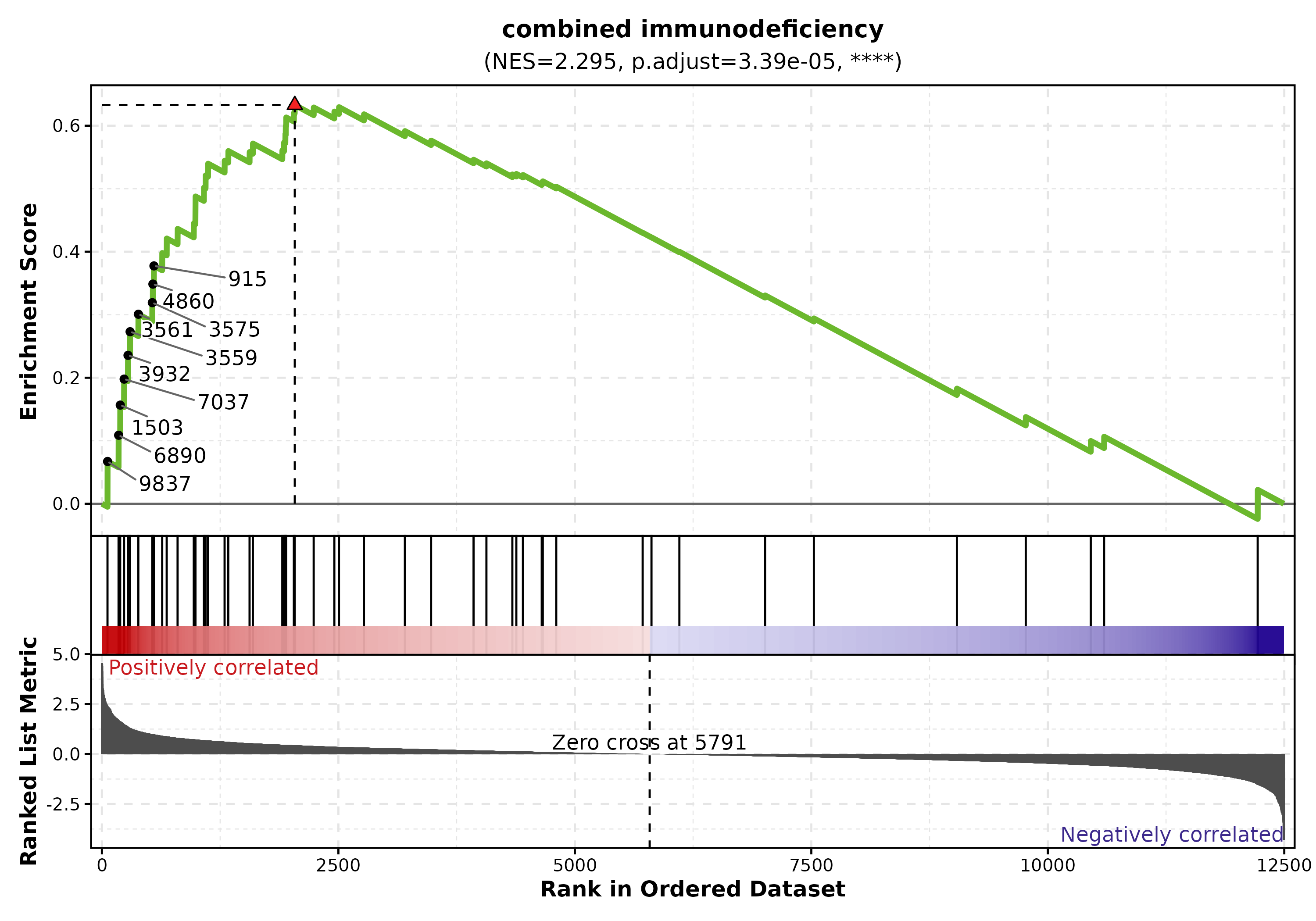
Category 3: Single-Cell & Spatial
Dimension Reduction
data("dim_example")
DimPlot(dim_example, dims = c("basis_1", "basis_2"),
group_by = "clusters", palette = "igv",
pt_size = 1.2, label = TRUE, label_insitu = TRUE,
title = "UMAP Cell Clustering")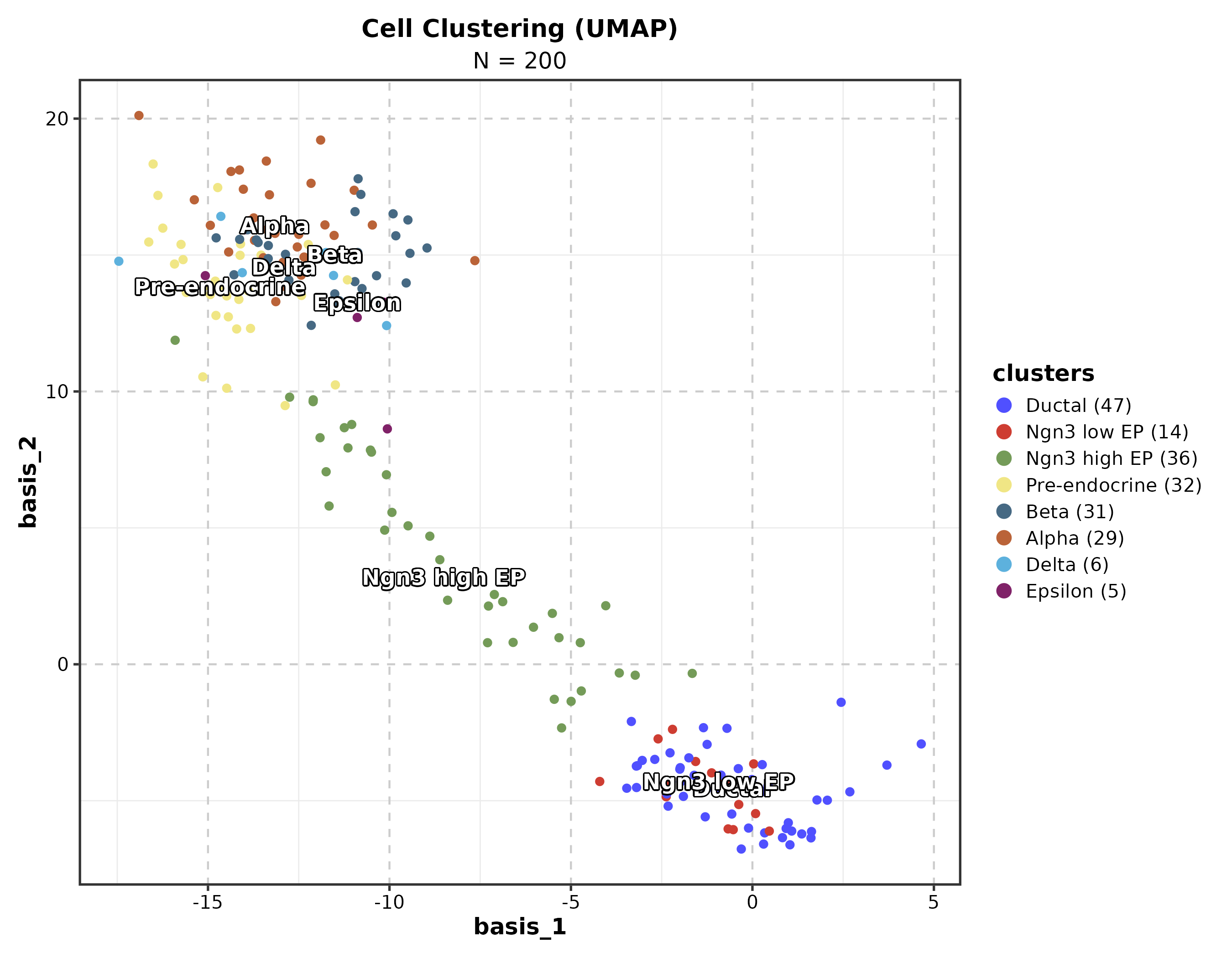
Feature Expression
dim_example$marker <- rnorm(nrow(dim_example))
FeatureDimPlot(dim_example, dims = c("basis_1", "basis_2"),
features = "marker", palette = "viridis", pt_size = 1.2,
title = "Feature Expression on UMAP")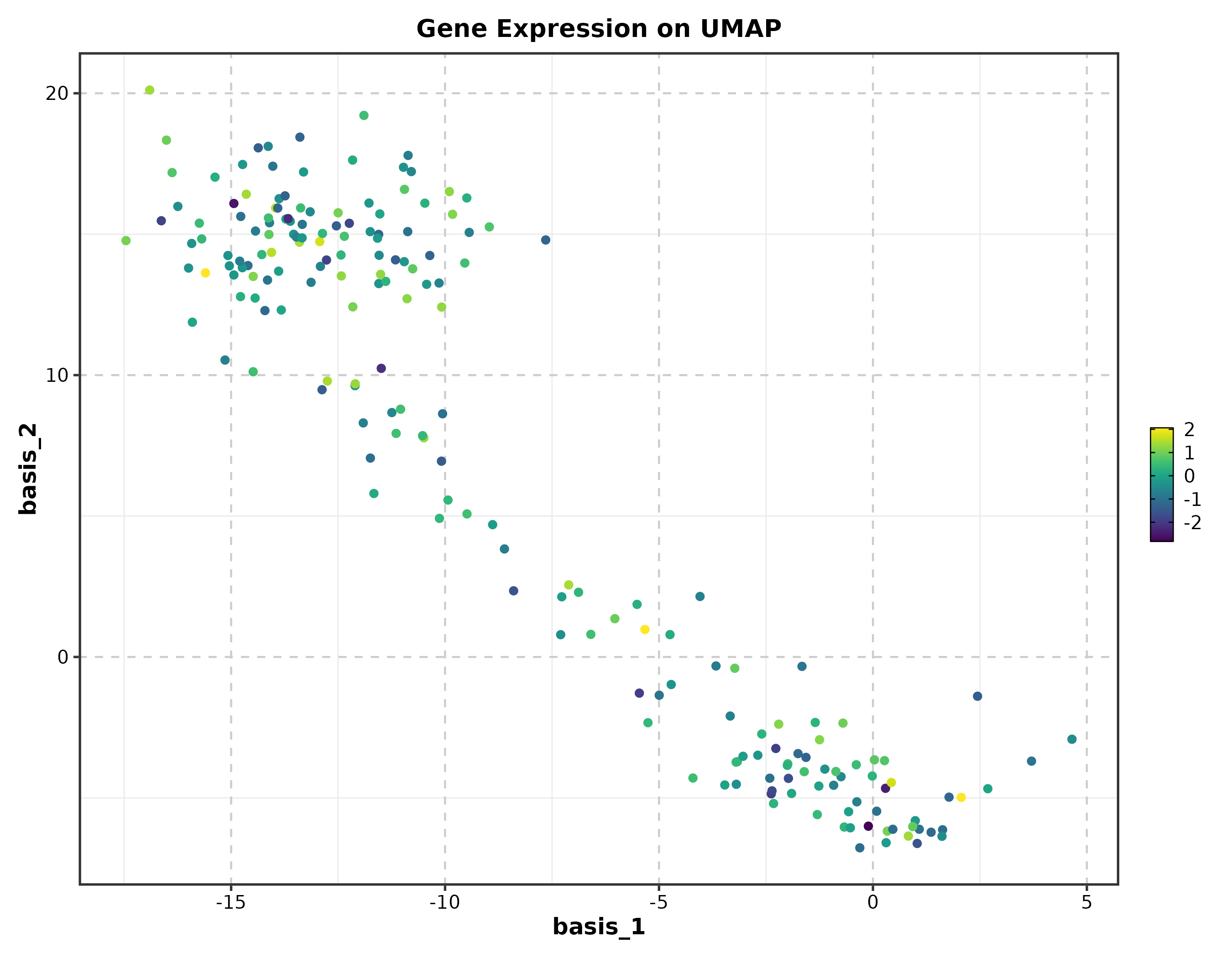
RNA Velocity
embedding <- as.matrix(dim_example[, c("basis_1", "basis_2")])
v_embedding <- as.matrix(dim_example[, c("stochasticbasis_1", "stochasticbasis_2")])
VelocityPlot(embedding = embedding, v_embedding = v_embedding,
plot_type = "grid", title = "RNA Velocity")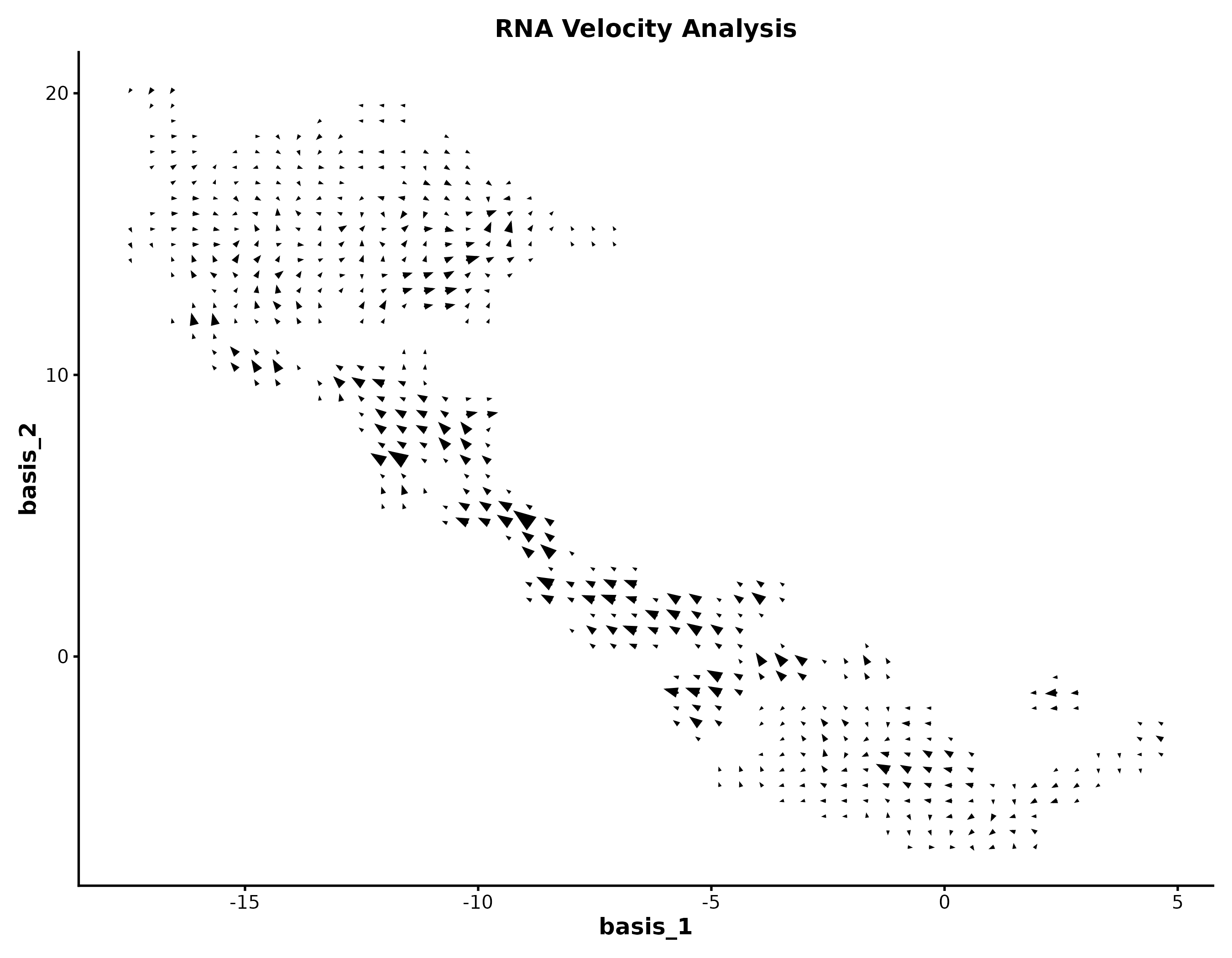
Category 4: Genomics
Volcano Plot
genes <- c("TP53","EGFR","MYC","KRAS","BRCA1","CDK4","PTEN","RB1",
"PIK3CA","AKT1","CD274","CTLA4","PDCD1","IDO1","VEGFA",
"BRAF","JAK2","STAT3","NRAS","HAVCR2")
deg <- data.frame(
gene = c(genes, paste0("Gene", 1:480)),
log2FC = c(rnorm(10, 2.5, 0.8), rnorm(10, -2.5, 0.8), rnorm(480, 0, 0.8)),
padj = c(runif(20, 1e-10, 1e-3), runif(480, 0.01, 1))
)
VolcanoPlot(deg, x = "log2FC", y = "padj", label_by = "gene",
x_cutoff = 1, y_cutoff = 0.05, nlabel = 10,
title = "Differential Expression")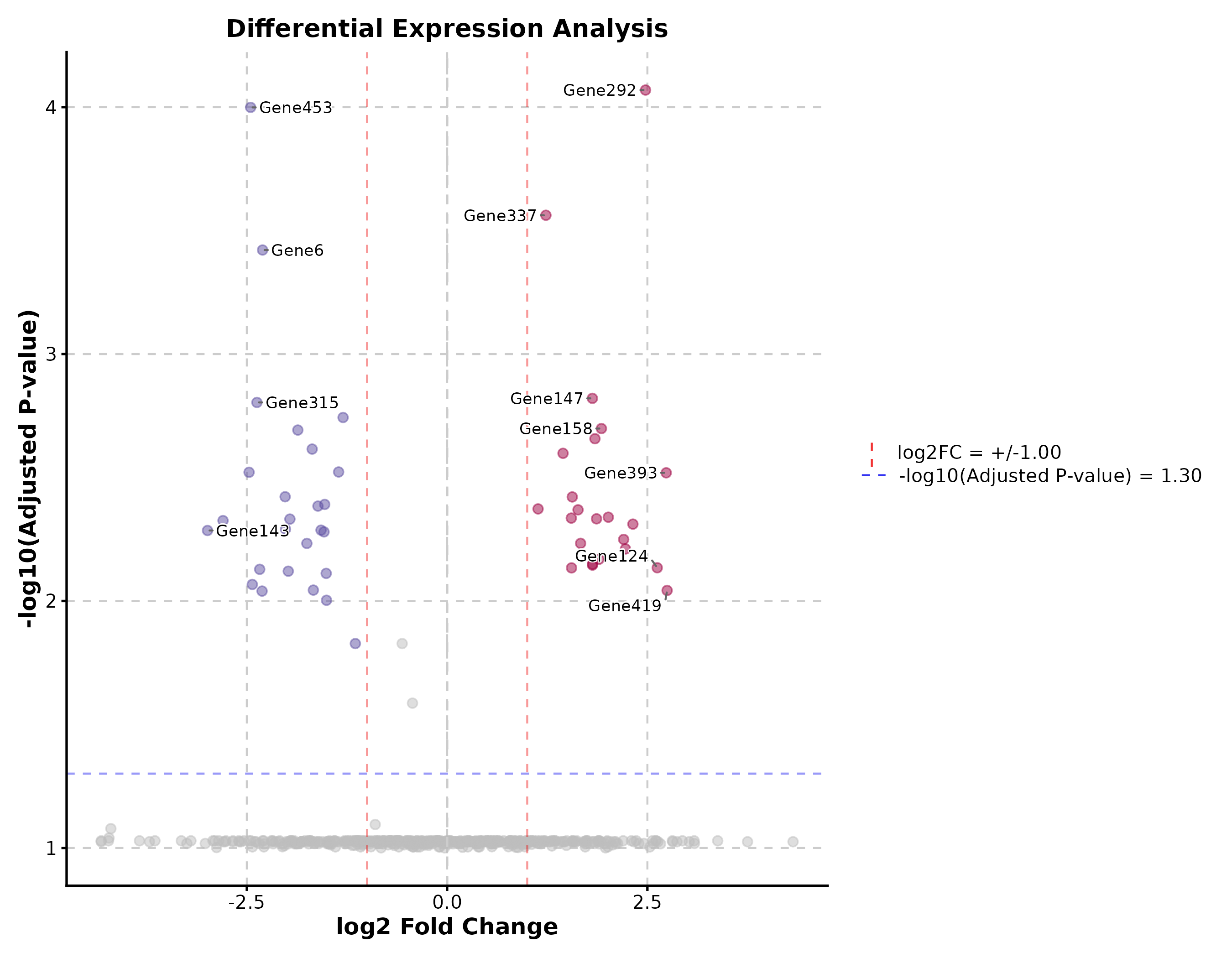
Manhattan Plot
gwas <- data.frame(
chr = rep(paste0("chr", 1:22), each = 500),
pos = rep(1:500, 22) * 1e5,
pvalue = runif(11000, 0, 1)
)
gwas$pvalue[sample(11000, 30)] <- runif(30, 0, 1e-8)
ManhattanPlot(gwas, chr_by = "chr", pos_by = "pos", pval_by = "pvalue",
signif = 5e-8, title = "GWAS Results")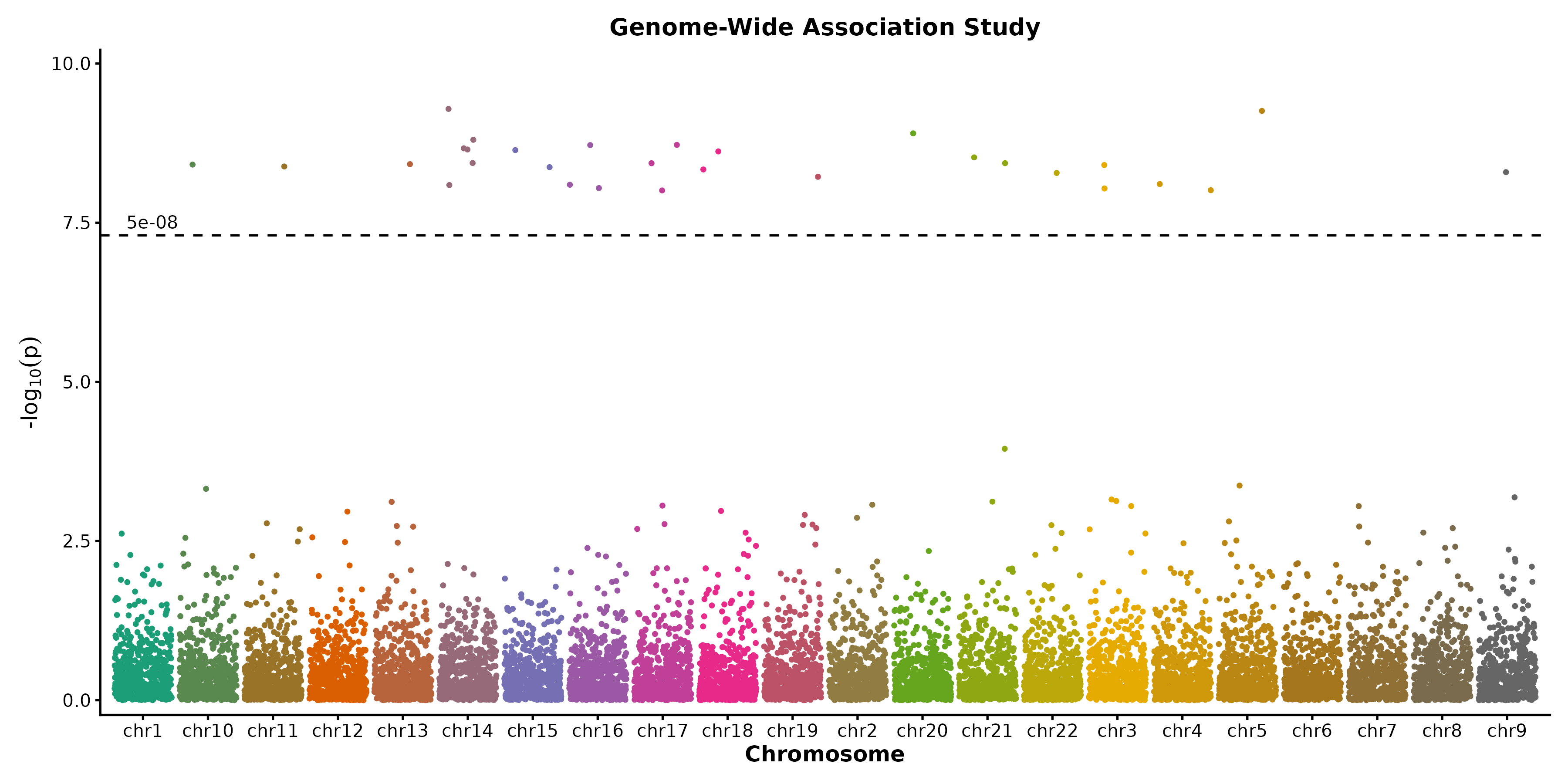
Category 5: Clinical & Prediction
Kaplan-Meier Curve
surv_data <- data.frame(
time = c(rexp(80, 0.008), rexp(70, 0.015)),
status = sample(0:1, 150, replace = TRUE, prob = c(0.35, 0.65)),
risk = rep(c("Low", "High"), c(80, 70))
)
KMPlot(surv_data, time = "time", status = "status", group_by = "risk",
palette = "jco", show_risk_table = TRUE, show_pval = TRUE,
title = "Overall Survival")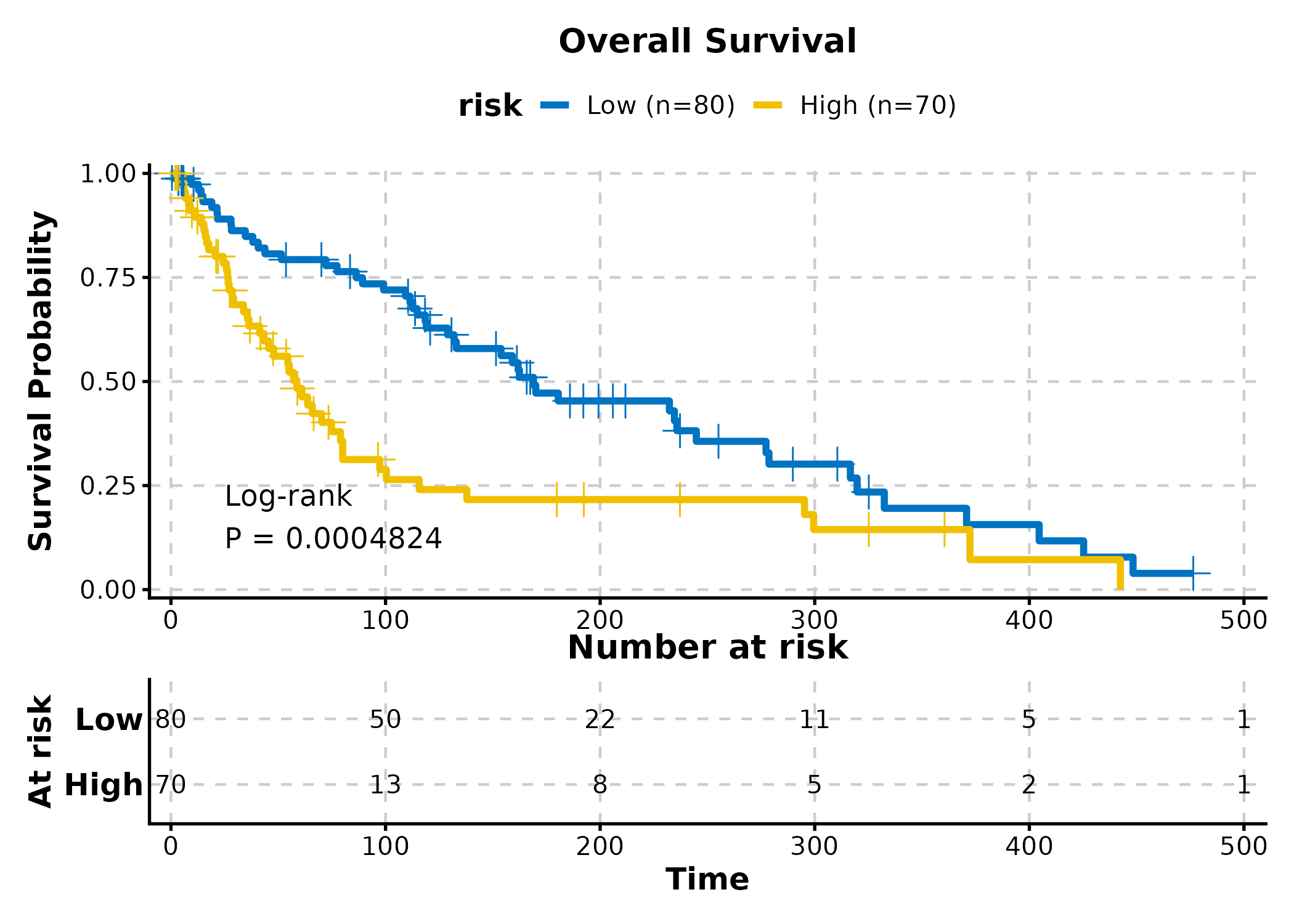
Cox Regression
cox_data <- data.frame(
time = rexp(200, 0.01),
event = sample(0:1, 200, replace = TRUE, prob = c(0.3, 0.7)),
age = rnorm(200, 60, 10),
gender = sample(c("Male", "Female"), 200, replace = TRUE),
stage = sample(c("I-II", "III-IV"), 200, replace = TRUE)
)
CoxPlot(cox_data, time = "time", event = "event",
vars = c("age", "gender", "stage"),
plot_type = "forest", palette = "nejm",
title = "Cox Forest Plot")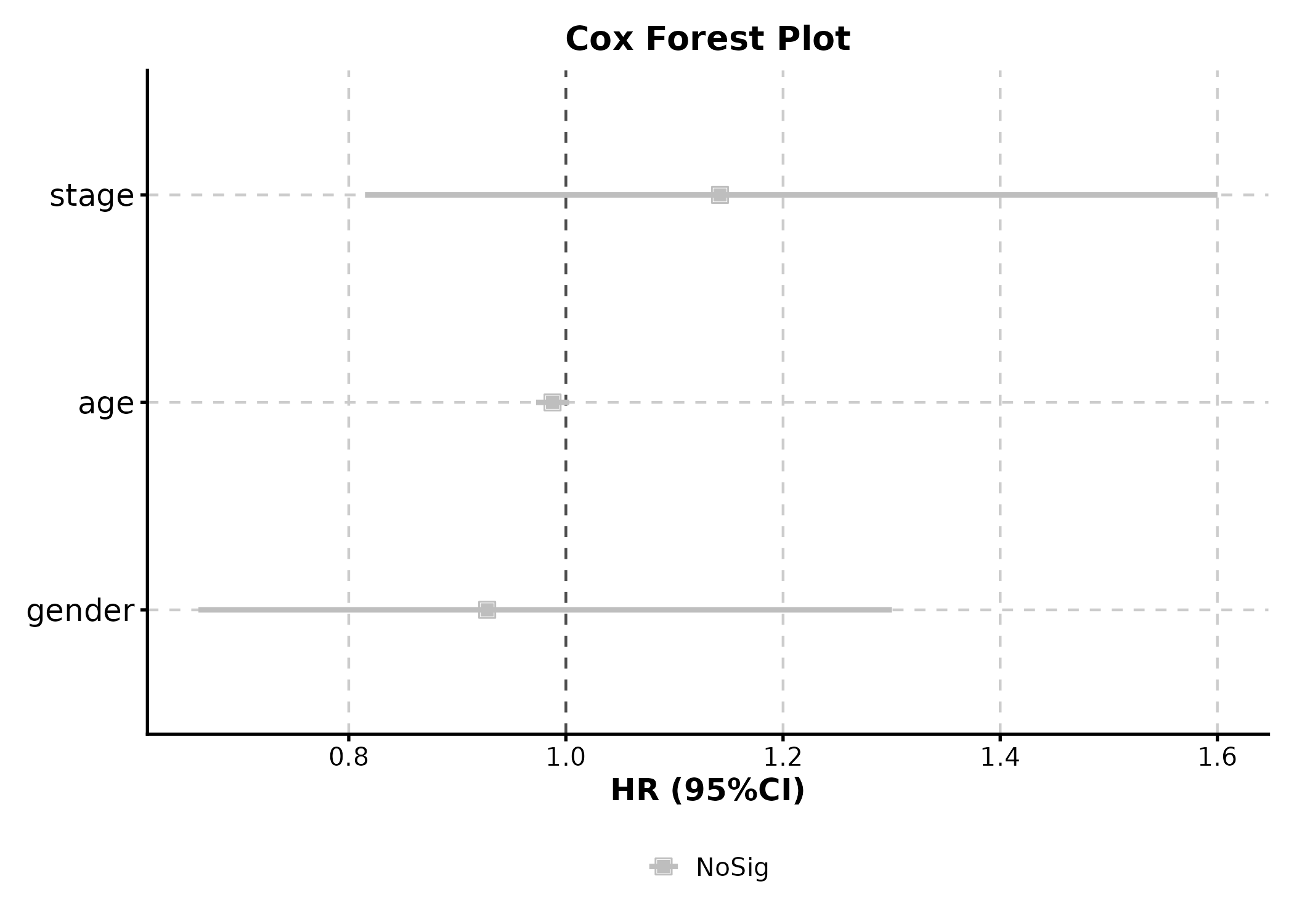
ROC Curve
roc_data <- data.frame(
truth = sample(0:1, 200, replace = TRUE),
score = rnorm(200)
)
roc_data$score <- roc_data$score + roc_data$truth * 1.5
ROCCurve(roc_data, truth_by = "truth", score_by = "score",
palette = "lancet", title = "ROC Analysis")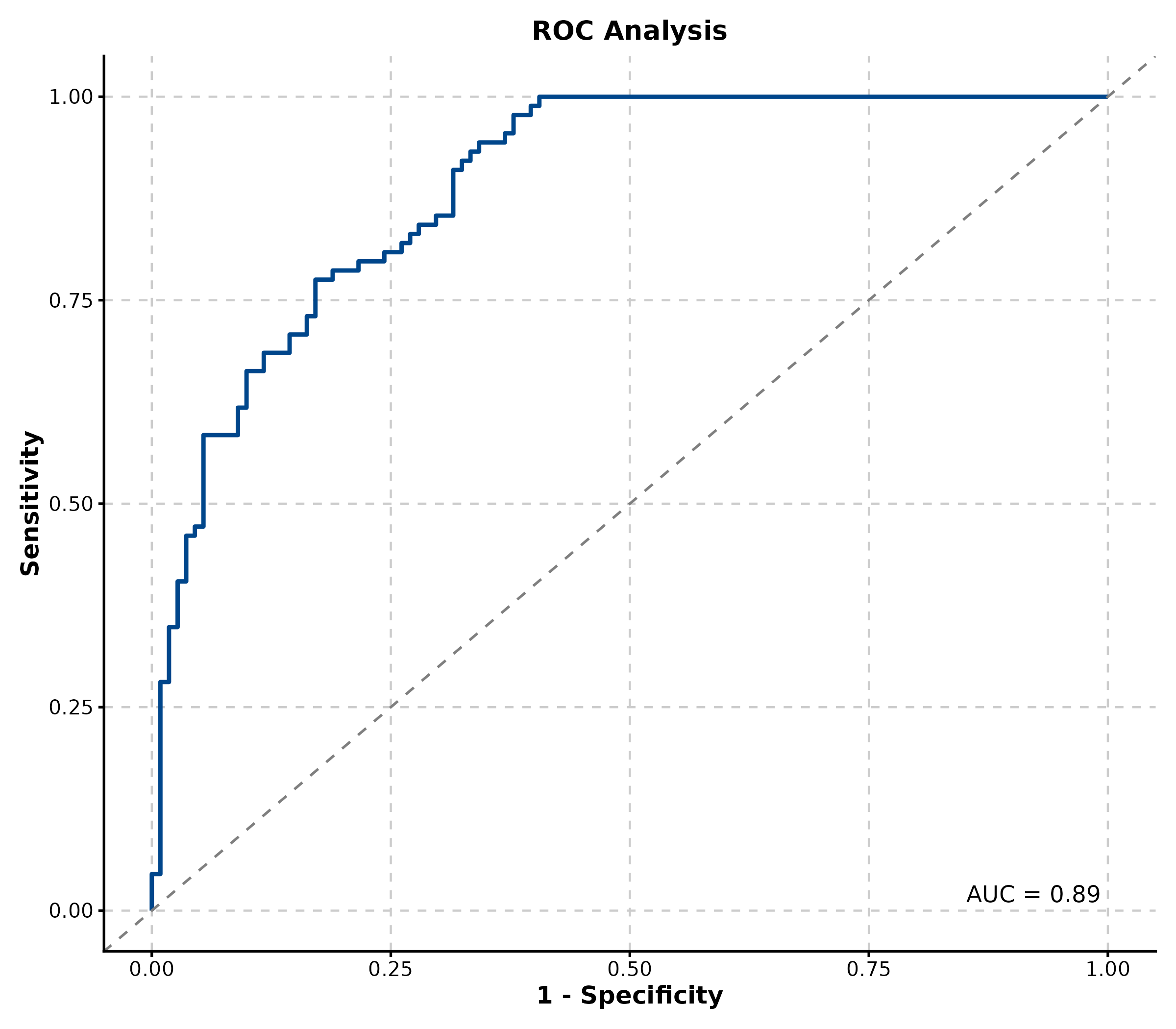
Decision Curve Analysis
New in v2.0.
dca_data <- data.frame(
truth = sample(0:1, 300, replace = TRUE),
model = runif(300)
)
dca_data$model <- ifelse(dca_data$truth == 1,
pmin(dca_data$model + 0.2, 1),
pmax(dca_data$model - 0.2, 0))
DecisionCurvePlot(dca_data, outcome = "truth", predictors = "model",
title = "Decision Curve Analysis")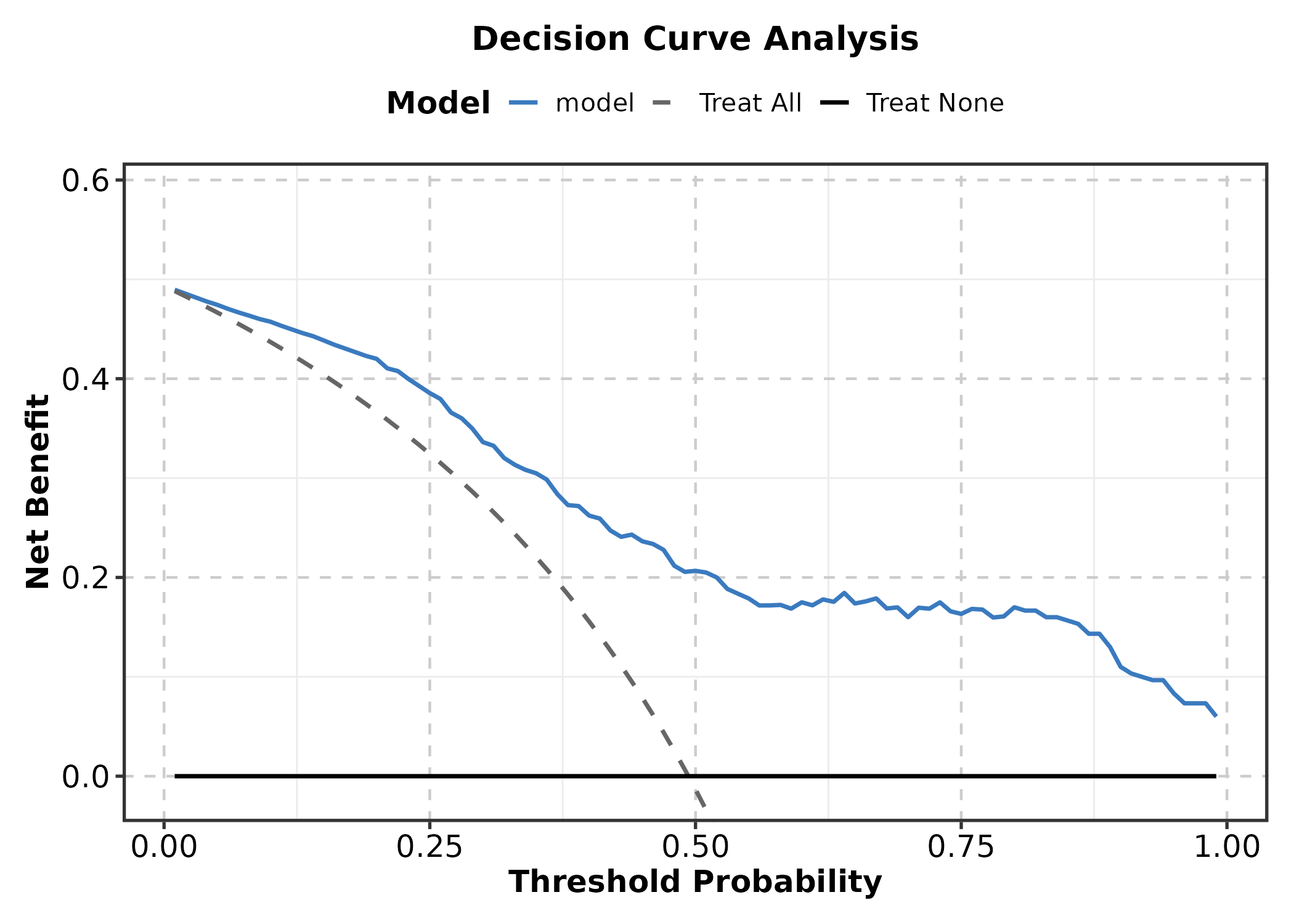
Category 6: Networks & Relationships
Heatmap
mat <- matrix(rnorm(120, 0, 1.5), nrow = 10,
dimnames = list(paste0("Gene", 1:10), paste0("Sample", 1:12)))
mat[1:4, 1:4] <- mat[1:4, 1:4] + 2
mat[5:7, 5:8] <- mat[5:7, 5:8] + 2
mat[8:10, 9:12] <- mat[8:10, 9:12] + 2
Heatmap(mat, palette = "RdBu",
show_row_names = TRUE, show_column_names = TRUE,
title = "Expression Heatmap")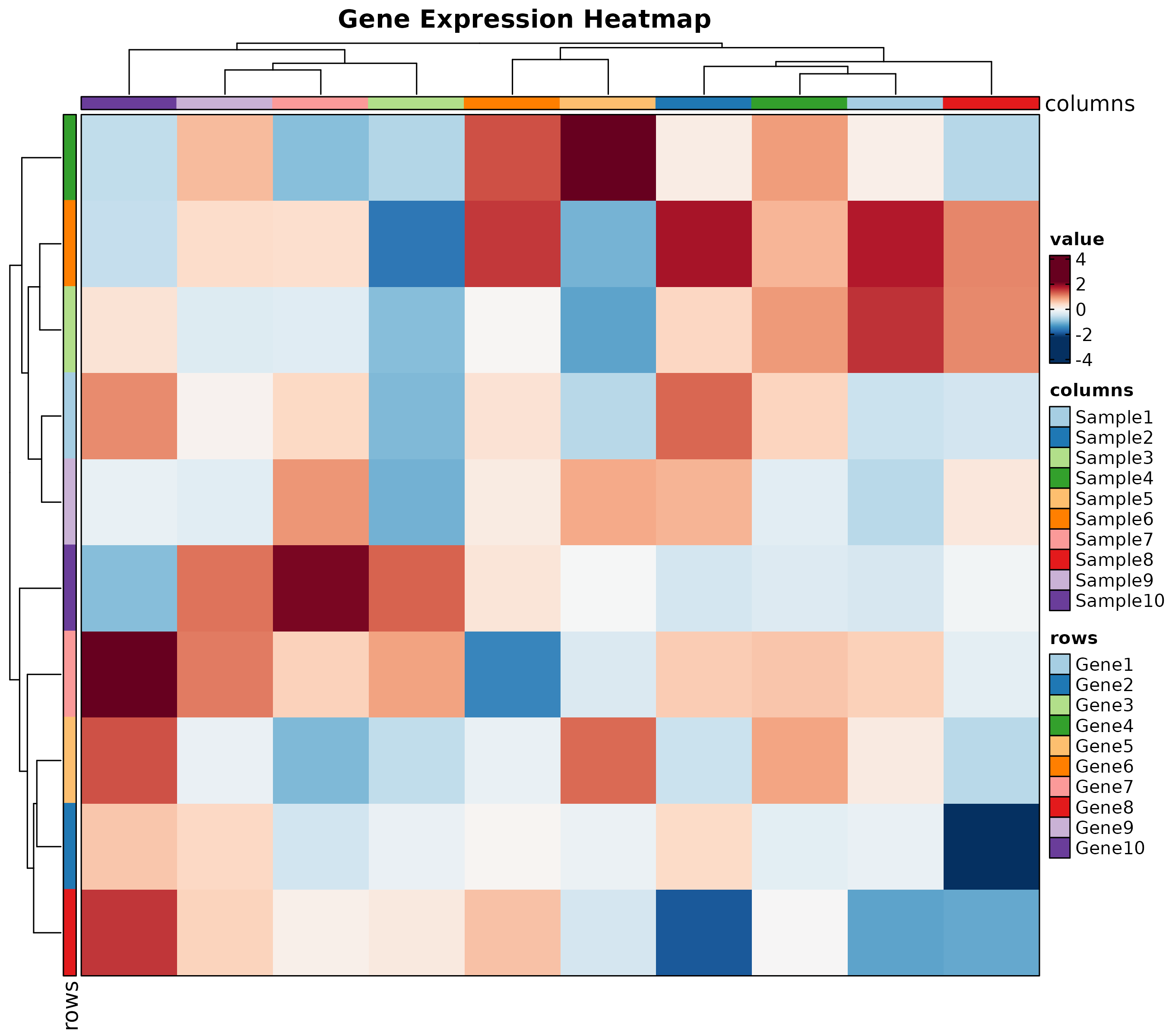
Chord Diagram
chord_data <- data.frame(
from = c("CD4 T", "CD8 T", "B cell", "NK", "Monocyte"),
to = c("Fibroblast", "Endothelial", "Fibroblast", "Tumor", "Tumor"),
value = c(15, 20, 10, 25, 18)
)
ChordPlot(chord_data, from = "from", to = "to", y = "value",
palette = "Set3", title = "Cell Interaction")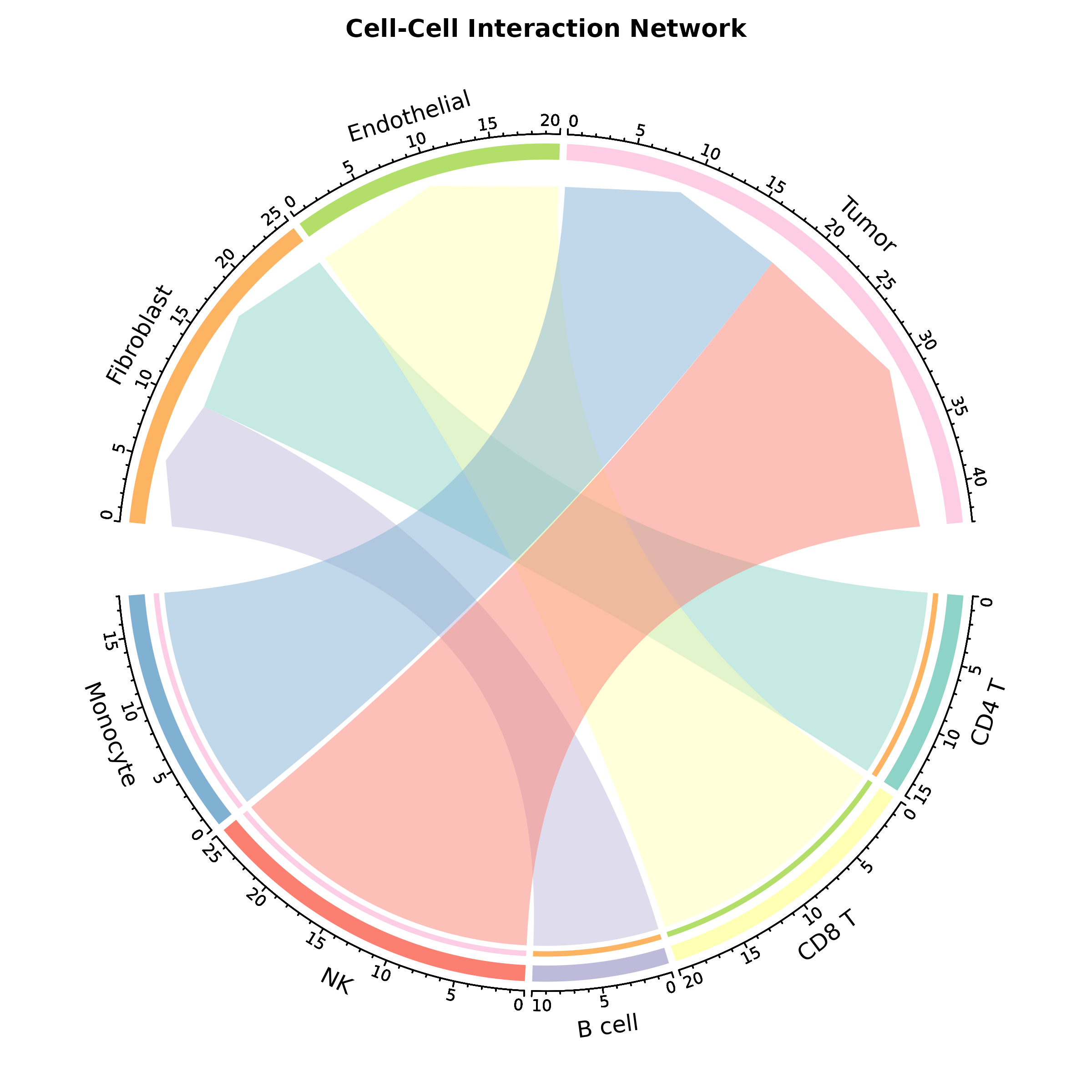
Sankey Plot
flow <- data.frame(
diagnosis = rep(c("Early", "Advanced", "Metastatic"), each = 3),
outcome = rep(c("Complete Response", "Partial Response", "Progressive"), 3),
n = c(40, 8, 2, 10, 20, 10, 2, 8, 20)
)
SankeyPlot(flow, x = c("diagnosis", "outcome"), y = "n",
links_fill_by = "diagnosis", palette = "Set3",
title = "Treatment Outcome Flow")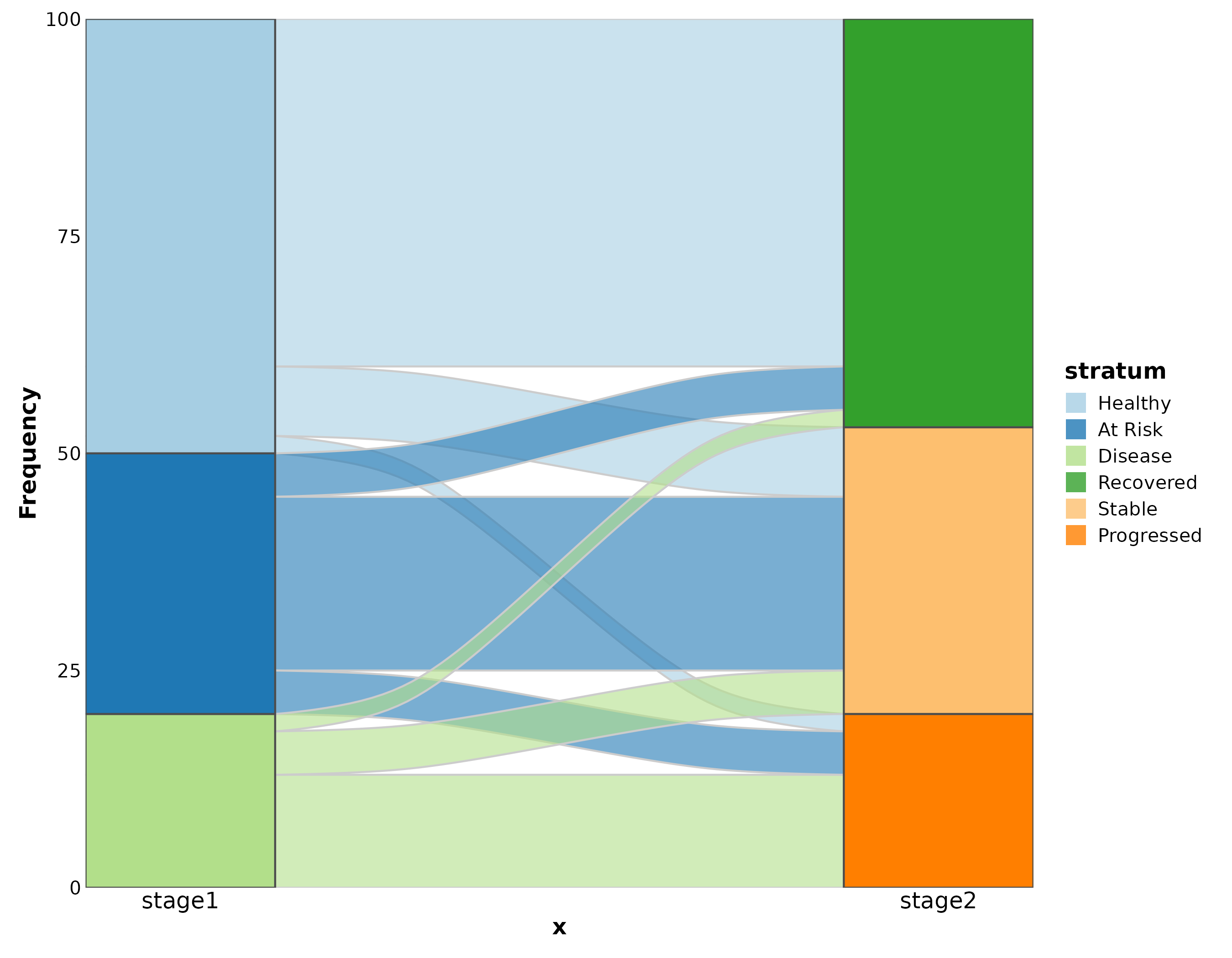
Category 7: Specialized
Dumbbell Plot
New in v2.0.
dumb <- data.frame(
gene = paste0("Gene", LETTERS[1:8]),
before = rnorm(8, 5, 1.5), after = rnorm(8, 8, 1.5)
)
DumbbellPlot(dumb, x_start = "before", x_end = "after", y = "gene",
palette = "Set1",
title = "Pre- vs Post-Treatment")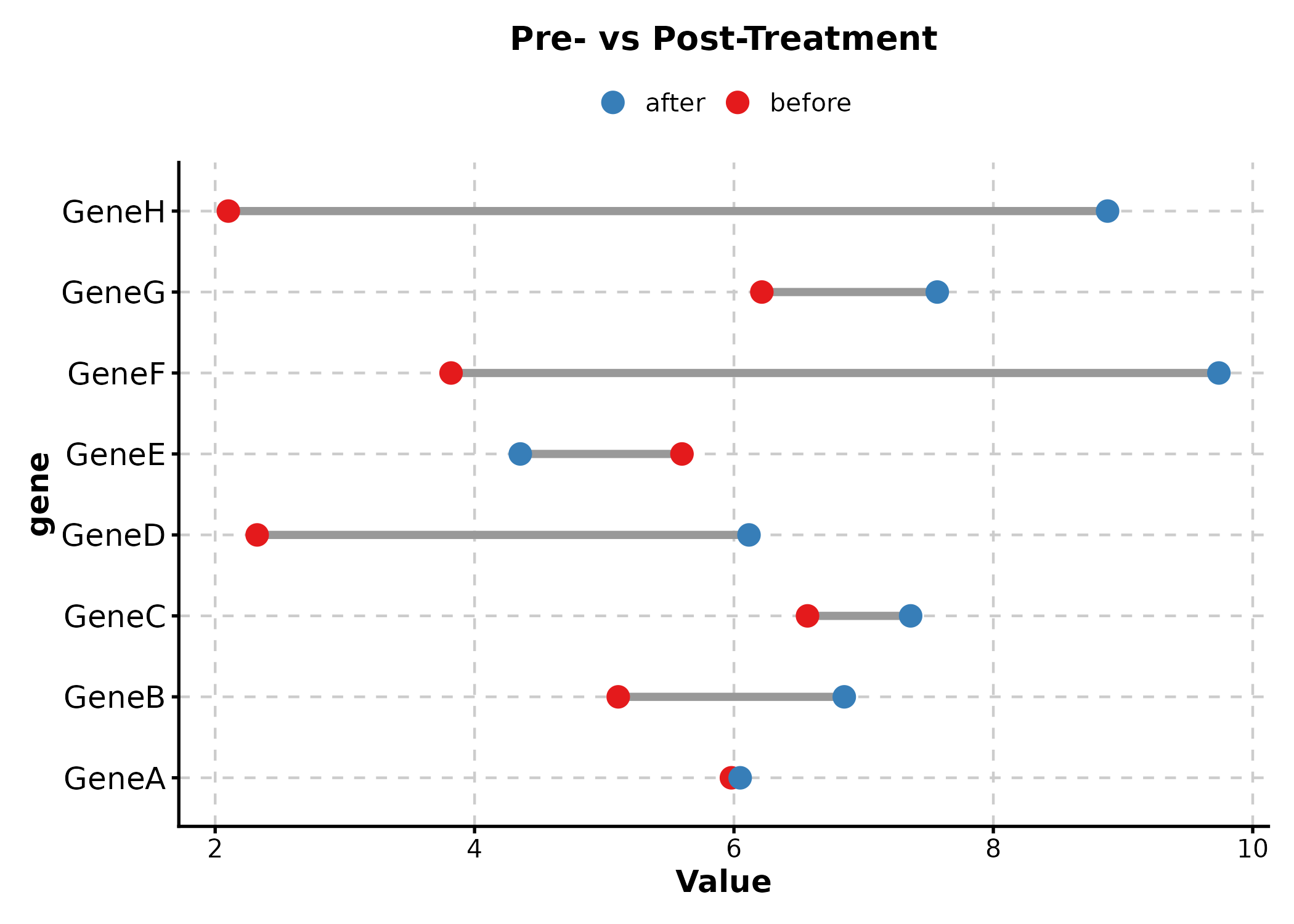
Waffle Plot
New in v2.0.
cell_comp <- data.frame(
type = c("T cell", "B cell", "NK", "Monocyte", "Other"),
count = c(35, 20, 15, 20, 10)
)
WafflePlot(cell_comp, x = "type", y = "count",
palette = "Set2", title = "Cell Proportions")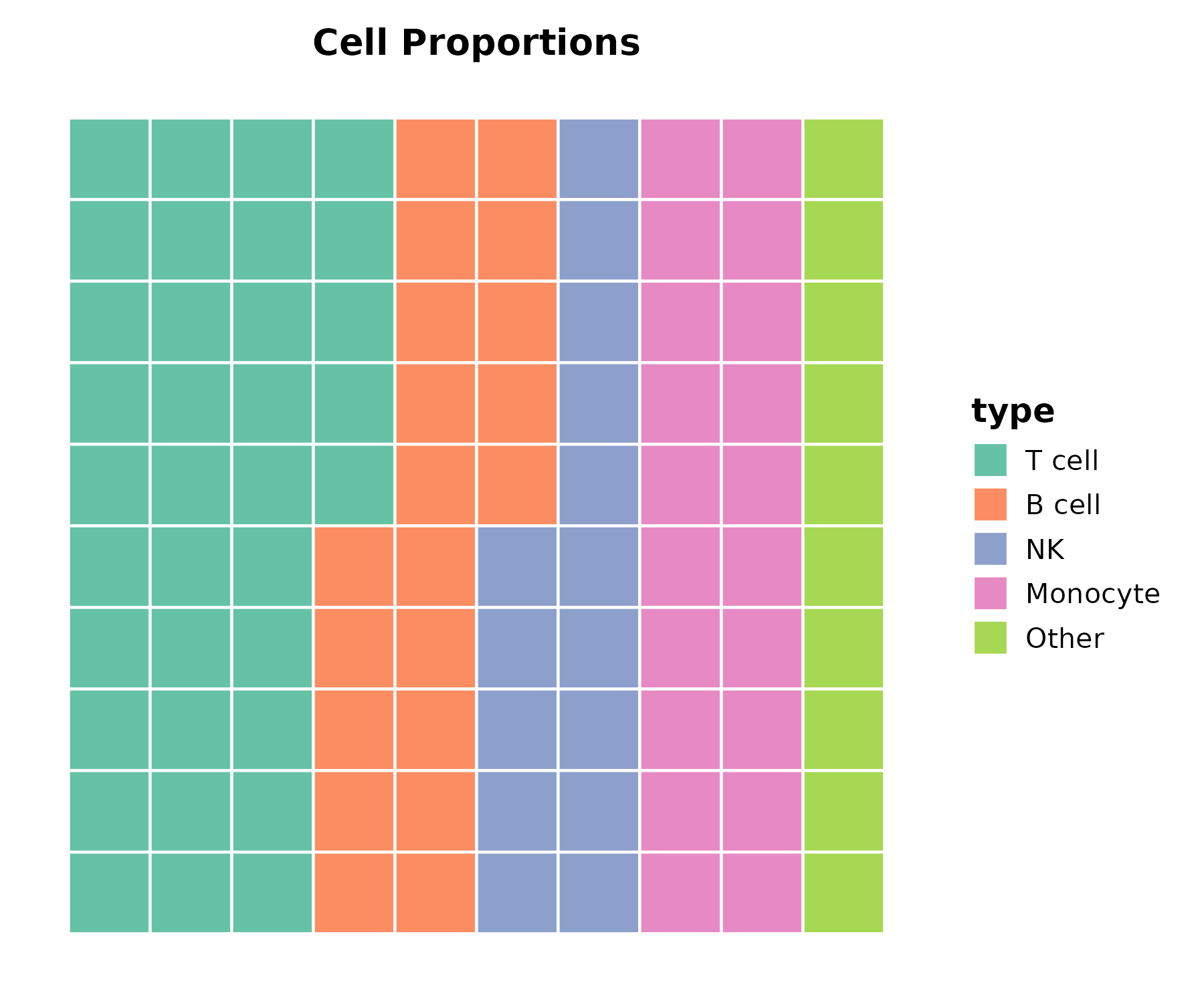
Timeline Plot
New in v2.0.
events <- data.frame(
event = c("Diagnosis", "Surgery", "Chemo", "Radiation", "Follow-up"),
start = as.Date(c("2024-01-15", "2024-02-10", "2024-03-01",
"2024-06-01", "2024-09-01")),
end = as.Date(c("2024-01-15", "2024-02-12", "2024-05-15",
"2024-07-15", "2024-12-01"))
)
TimelinePlot(events, start = "start", end = "end", label = "event",
palette = "Set2", title = "Treatment Timeline")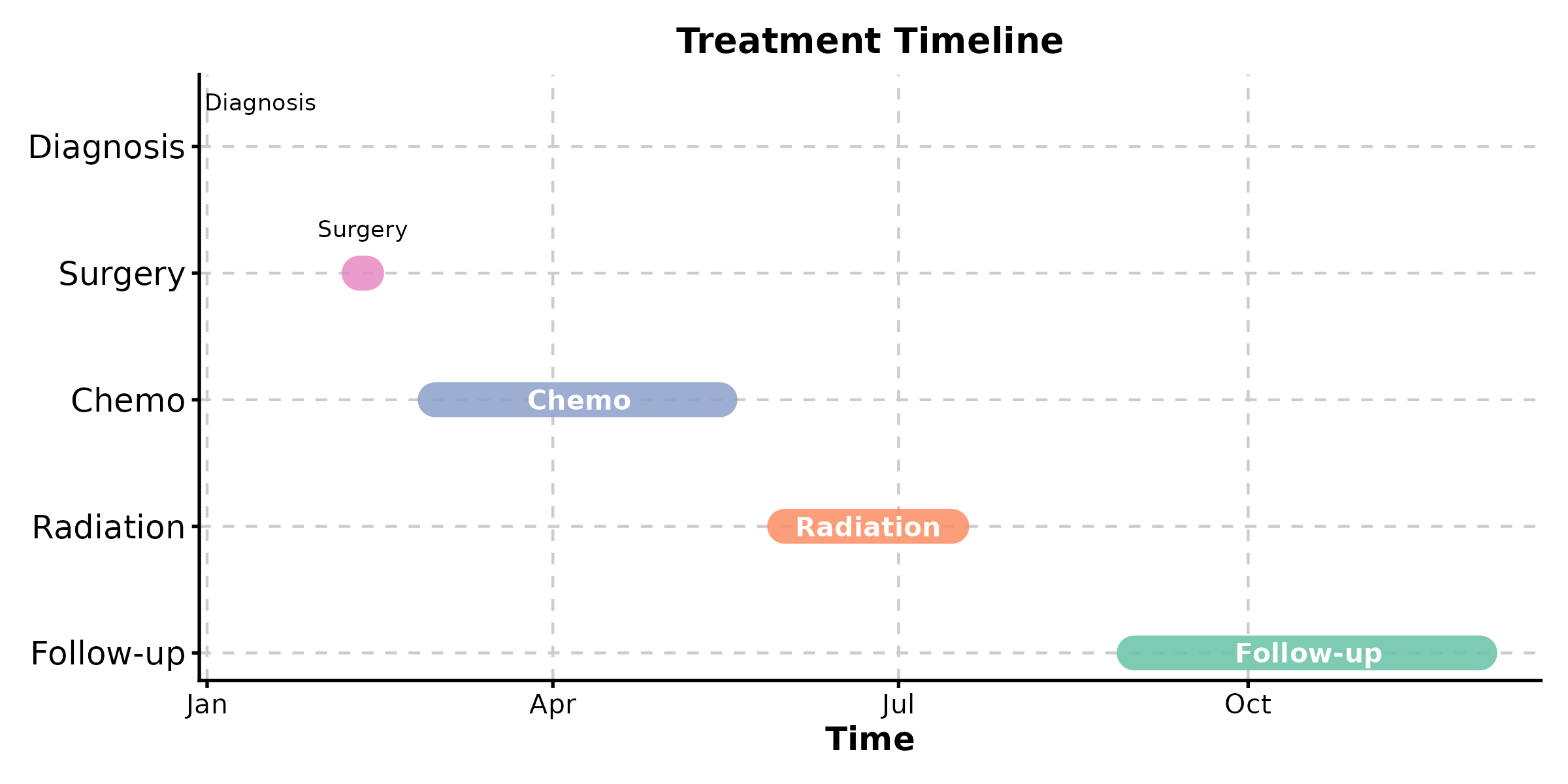
Category 8: Earth & Environmental
New in v2.0.
Contour Plot
grid <- expand.grid(
x = seq(-3, 3, length.out = 50),
y = seq(-3, 3, length.out = 50)
)
grid$z <- with(grid, sin(x) * cos(y) * exp(-(x^2 + y^2) / 8))
ContourPlot(grid, x = "x", y = "y", z = "z",
type = "filled", palette = "Spectral",
title = "Scalar Field")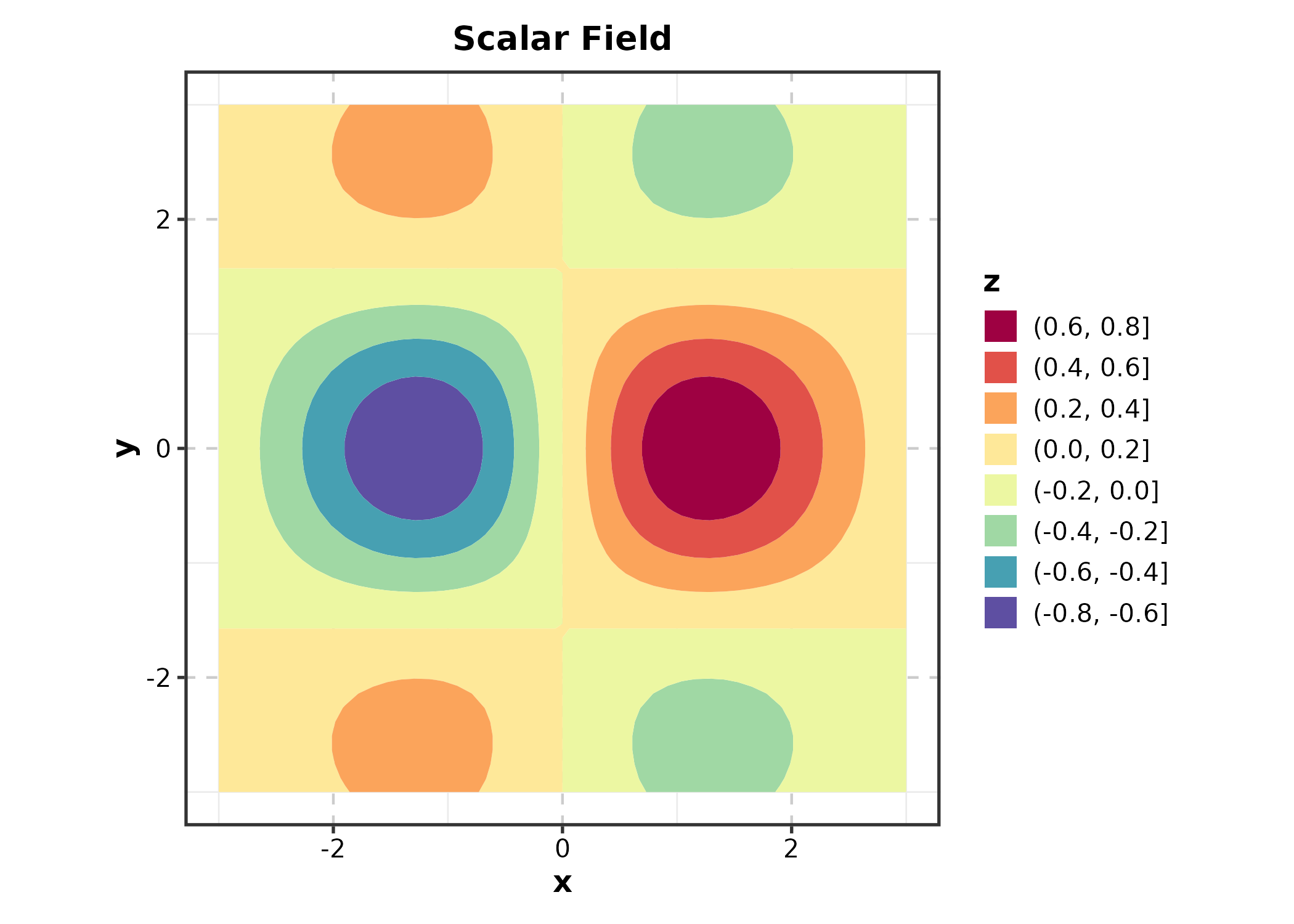
Category 9: Meta-Analysis & Agreement
New in v2.0.
Forest Plot
meta <- data.frame(
study = c("Study A", "Study B", "Study C", "Study D", "Overall"),
or = c(1.32, 0.85, 1.51, 1.08, 1.15),
lower = c(0.95, 0.60, 1.05, 0.78, 0.95),
upper = c(1.84, 1.20, 2.17, 1.50, 1.38),
weight = c(25, 20, 18, 22, NA),
is_summary = c(FALSE, FALSE, FALSE, FALSE, TRUE)
)
ForestPlot(meta, estimate = "or", ci_lower = "lower", ci_upper = "upper",
label = "study", weight = "weight", is_summary = "is_summary",
null_value = 1, title = "Meta-Analysis")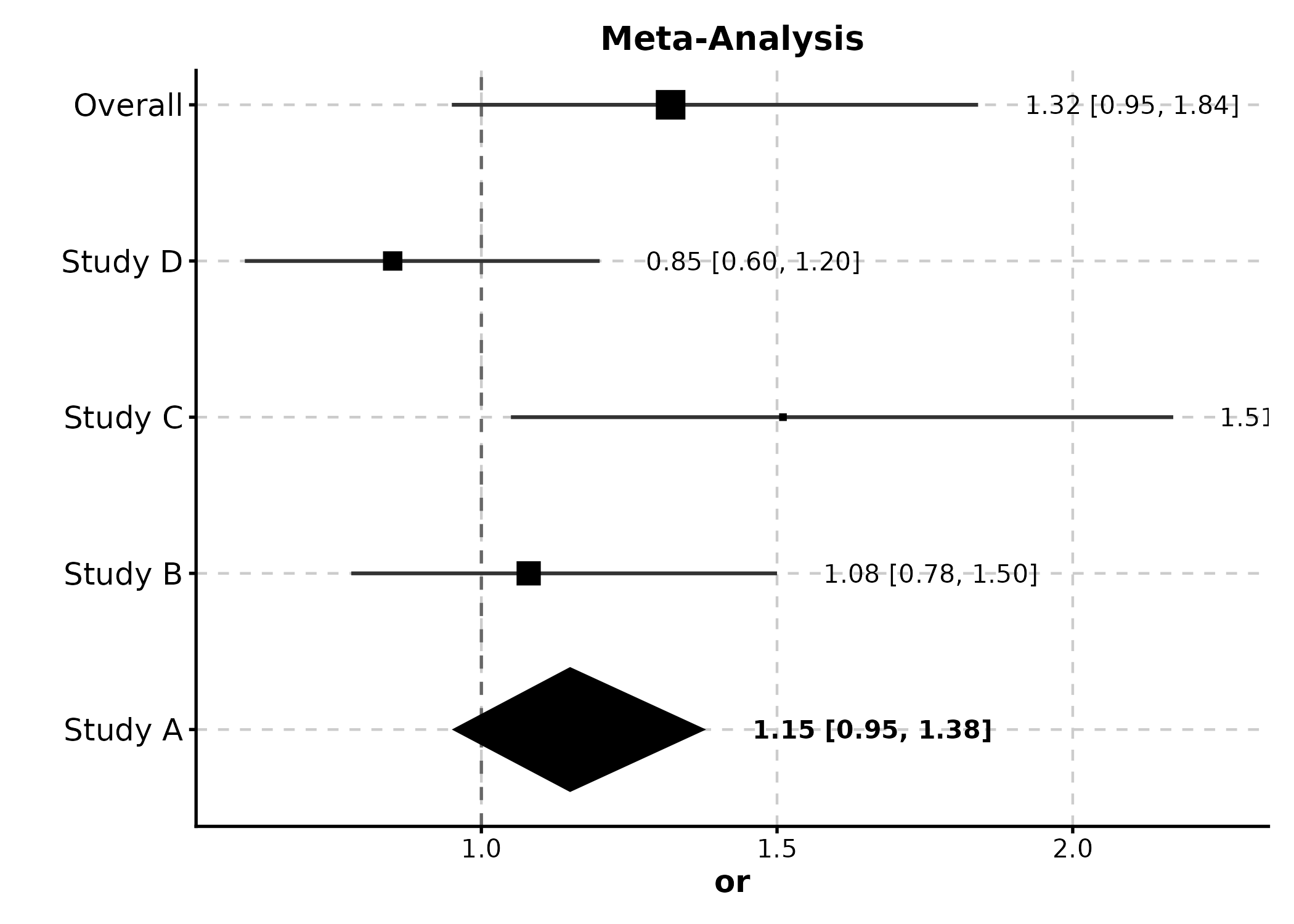
Bland-Altman Plot
agree <- data.frame(
method_A = rnorm(50, 100, 15),
method_B = rnorm(50, 100, 15)
)
agree$method_B <- agree$method_A + rnorm(50, 2, 5)
BlandAltmanPlot(agree, method1 = "method_A", method2 = "method_B",
title = "Method Agreement")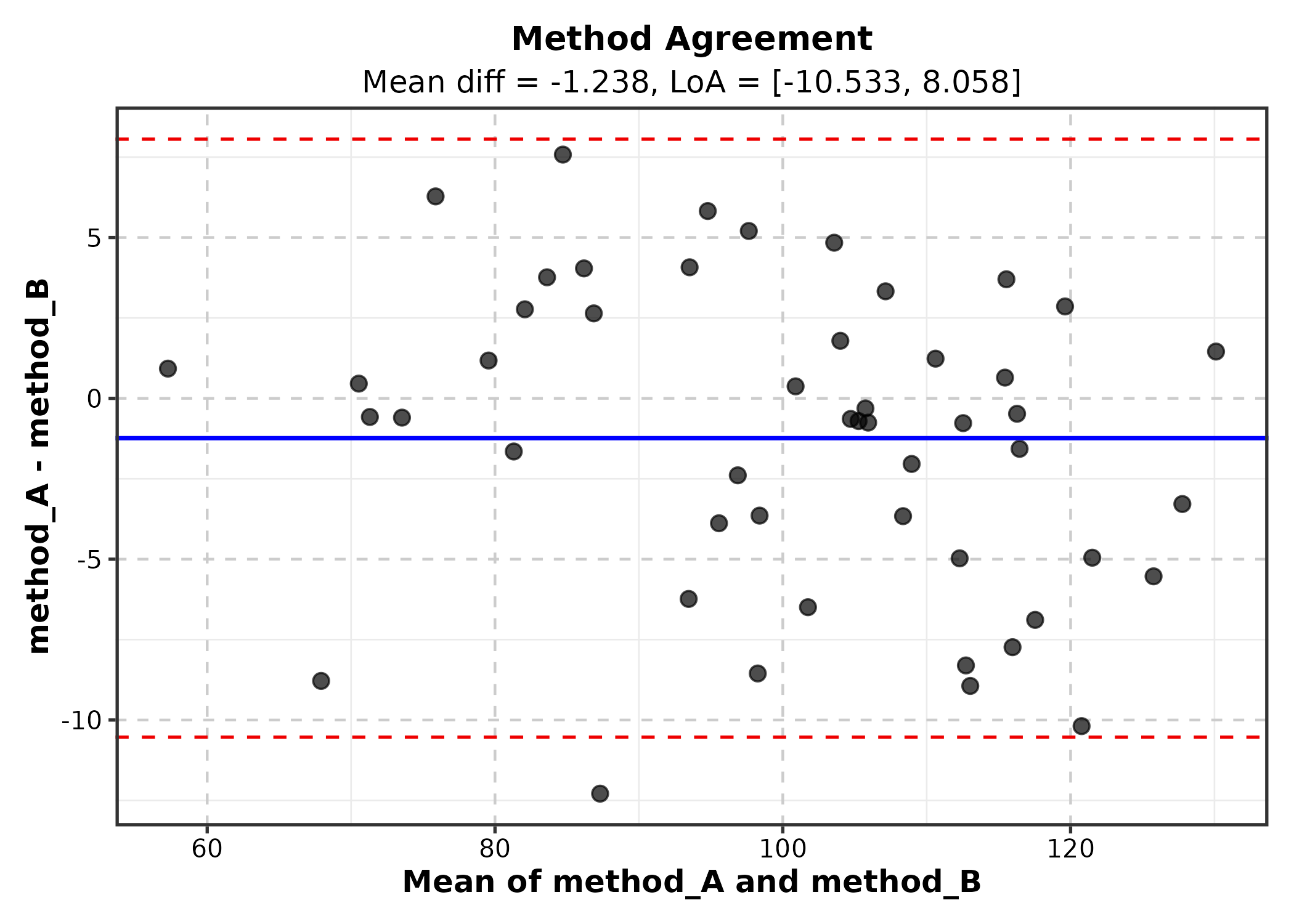
Category 10: Ecology & Evolution
New in v2.0.
Ordination Plot
Requires pre-computed ordination coordinates.
library(vegan)
ord <- metaMDS(sp_mat, k = 2, trace = 0)
ord_df <- as.data.frame(scores(ord, display = "sites"))
ord_df$habitat <- env$habitat
OrdinationPlot(ord_df, x = "NMDS1", y = "NMDS2",
group_by = "habitat", palette = "Set1",
title = "Community Ordination")Phylogenetic Tree
tree <- ape::rtree(12, tip.label = paste0("Species_", LETTERS[1:12]))
PhyloTreePlot(tree, palette = "Set2",
title = "Phylogenetic Tree")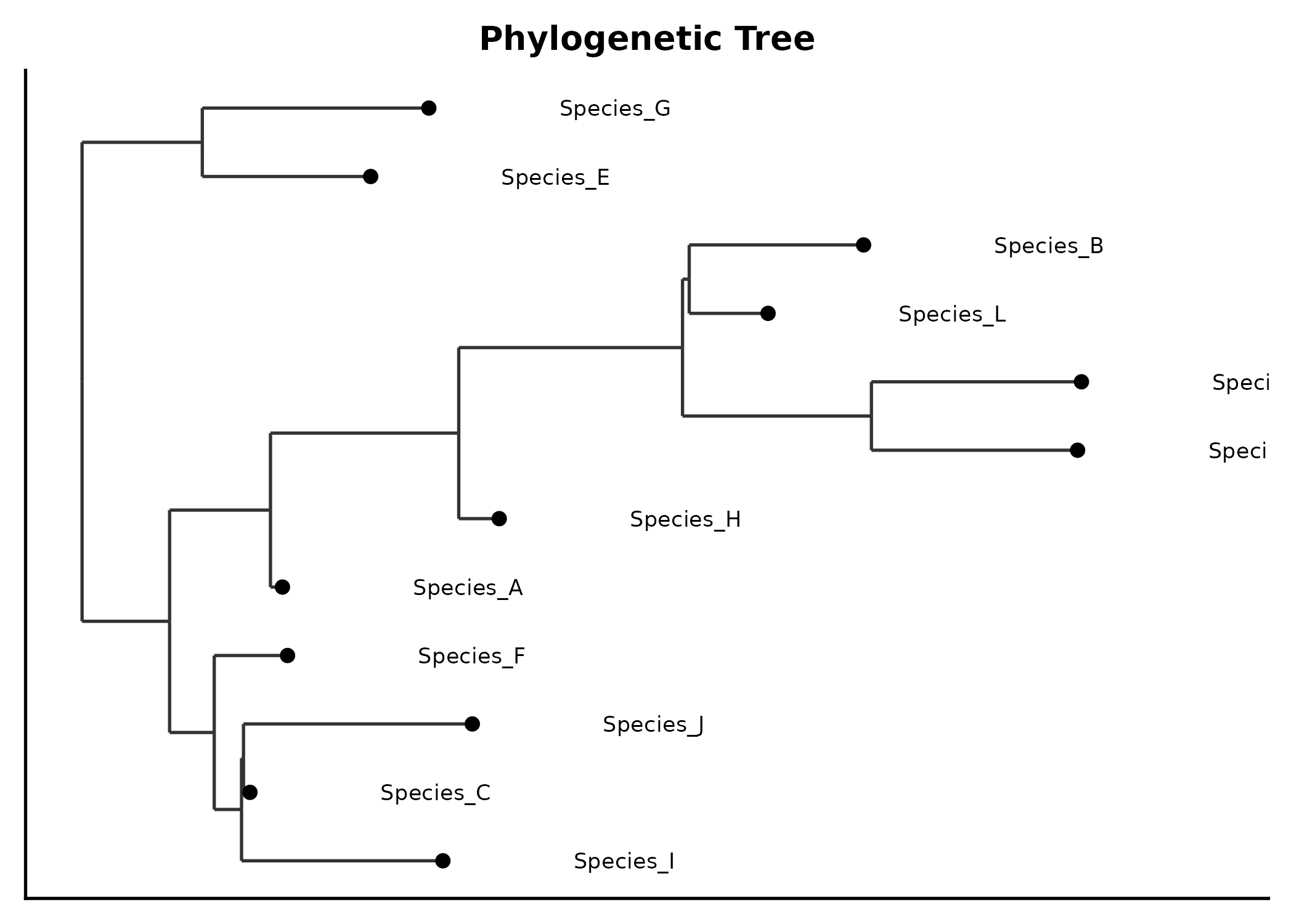
Category 11: Physics & Engineering
New in v2.0.
Quiver Plot
field <- expand.grid(x = seq(-2, 2, 0.4), y = seq(-2, 2, 0.4))
field$u <- -field$y
field$v <- field$x
QuiverPlot(field, x = "x", y = "y", u = "u", v = "v",
title = "Vector Field")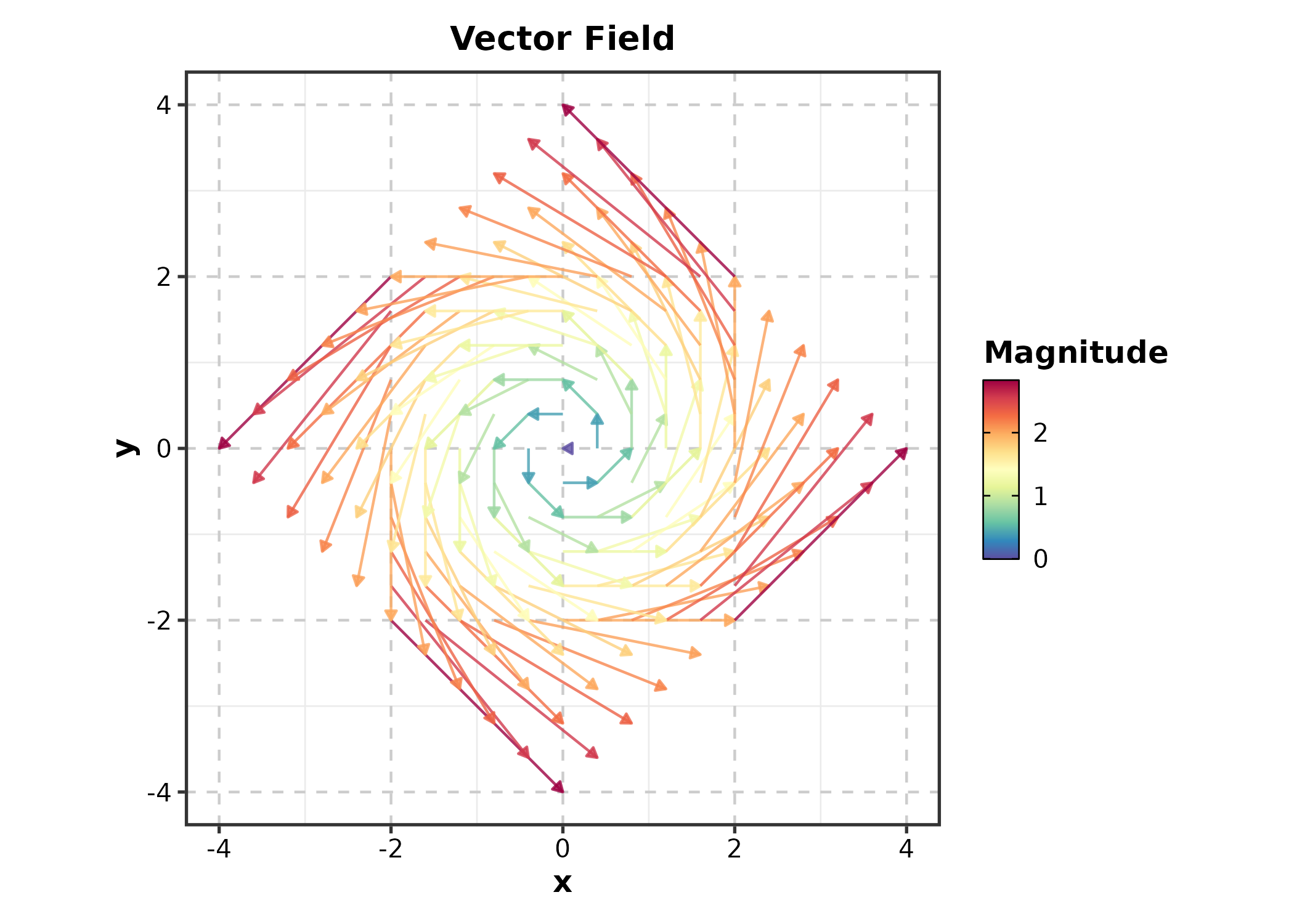
Category 13: Machine Learning
New in v2.0.
Confusion Matrix
set.seed(42)
pred <- data.frame(
actual = sample(c("Positive", "Negative"), 200,
replace = TRUE, prob = c(0.4, 0.6)),
predicted = sample(c("Positive", "Negative"), 200,
replace = TRUE, prob = c(0.45, 0.55))
)
correct <- sample(200, 150)
pred$predicted[correct] <- pred$actual[correct]
ConfusionMatrixPlot(pred, truth = "actual", predicted = "predicted",
palette = "Blues", title = "Classification Result")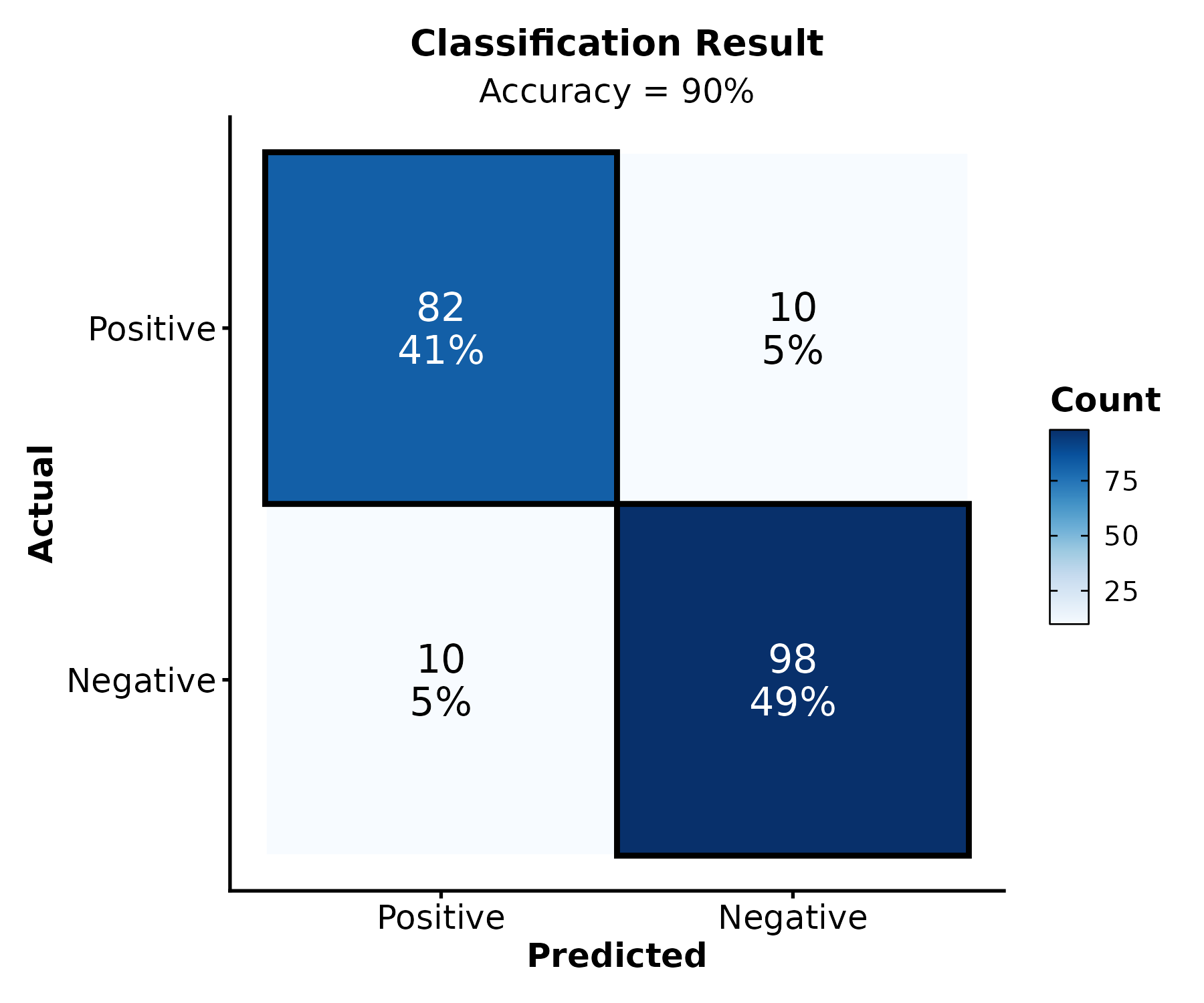
Discovering Functions
Use ggforge_gallery() to browse available functions by
category:
ggforge_gallery()
ggforge_gallery("enrichment")
ggforge_gallery("survival")Getting Help
-
Documentation:
?FunctionNameorhelp(FunctionName) -
Examples:
example(FunctionName) - GitHub Issues: https://github.com/Zaoqu-Liu/ggforge/issues
Acknowledgments
ggforge is built upon plotthis by Panwen Wang. Thanks also to ggplot2 and the R community.
Session Info
sessionInfo()
#> R version 4.6.0 (2026-04-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] dplyr_1.2.1 ggforge_2.0.1 ggplot2_4.0.3
#>
#> loaded via a namespace (and not attached):
#> [1] gridExtra_2.3 rlang_1.2.0 magrittr_2.0.5
#> [4] clue_0.3-68 GetoptLong_1.1.1 otel_0.2.0
#> [7] matrixStats_1.5.0 compiler_4.6.0 mgcv_1.9-4
#> [10] png_0.1-9 systemfonts_1.3.2 vctrs_0.7.3
#> [13] ggalluvial_0.12.6 stringr_1.6.0 pkgconfig_2.0.3
#> [16] shape_1.4.6.1 crayon_1.5.3 fastmap_1.2.0
#> [19] magick_2.9.1 backports_1.5.1 labeling_0.4.3
#> [22] ggraph_2.2.2 rmarkdown_2.31 ragg_1.5.2
#> [25] purrr_1.2.2 xfun_0.58 cachem_1.1.0
#> [28] jsonlite_2.0.0 tweenr_2.0.3 broom_1.0.13
#> [31] parallel_4.6.0 cluster_2.1.8.2 R6_2.6.1
#> [34] bslib_0.11.0 stringi_1.8.7 RColorBrewer_1.1-3
#> [37] car_3.1-5 jquerylib_0.1.4 ggmanh_1.16.0
#> [40] Rcpp_1.1.1-1.1 iterators_1.0.14 knitr_1.51
#> [43] zoo_1.8-15 IRanges_2.46.0 Matrix_1.7-5
#> [46] splines_4.6.0 igraph_2.3.2 tidyselect_1.2.1
#> [49] abind_1.4-8 dichromat_2.0-0.1 yaml_2.3.12
#> [52] viridis_0.6.5 doParallel_1.0.17 codetools_0.2-20
#> [55] plyr_1.8.9 lattice_0.22-9 tibble_3.3.1
#> [58] withr_3.0.2 S7_0.2.2 evaluate_1.0.5
#> [61] gridGraphics_0.5-1 desc_1.4.3 survival_3.8-6
#> [64] isoband_0.3.0 polyclip_1.10-7 circlize_0.4.18
#> [67] pillar_1.11.1 ggpubr_0.6.3 carData_3.0-6
#> [70] checkmate_2.3.4 foreach_1.5.2 stats4_4.6.0
#> [73] generics_0.1.4 metR_0.18.3 S4Vectors_0.50.1
#> [76] scales_1.4.0 glue_1.8.1 proxyC_0.5.2
#> [79] tools_4.6.0 ggnewscale_0.5.2 data.table_1.18.4
#> [82] ggsignif_0.6.4 forcats_1.0.1 fs_2.1.0
#> [85] graphlayouts_1.2.3 Cairo_1.7-0 tidygraph_1.3.1
#> [88] grid_4.6.0 tidyr_1.3.2 ape_5.8-1
#> [91] colorspace_2.1-2 nlme_3.1-169 patchwork_1.3.2
#> [94] ggforce_0.5.0 Formula_1.2-5 cli_3.6.6
#> [97] textshaping_1.0.5 plotROC_2.3.3 viridisLite_0.4.3
#> [100] ComplexHeatmap_2.28.0 gtable_0.3.6 rstatix_0.7.3
#> [103] sass_0.4.10 digest_0.6.39 BiocGenerics_0.58.1
#> [106] ggrepel_0.9.8 rjson_0.2.23 htmlwidgets_1.6.4
#> [109] farver_2.1.2 memoise_2.0.1 htmltools_0.5.9
#> [112] pkgdown_2.2.0 lifecycle_1.0.5 GlobalOptions_0.1.4
#> [115] MASS_7.3-65