This tutorial demonstrates every plotting function in ggforge (77
total) with practical examples. Functions requiring optional packages
that may not be available use eval=FALSE.
1. Statistics
BoxPlot
treat <- data.frame(
group = rep(c("Control", "Low Dose", "High Dose"), each = 35),
response = c(rnorm(35, 5, 1.5), rnorm(35, 7, 2), rnorm(35, 10, 2)),
sex = sample(c("Male", "Female"), 105, replace = TRUE)
)
BoxPlot(treat, x = "group", y = "response", palette = "lancet",
add_point = TRUE, pt_alpha = 0.3,
title = "Basic Box Plot")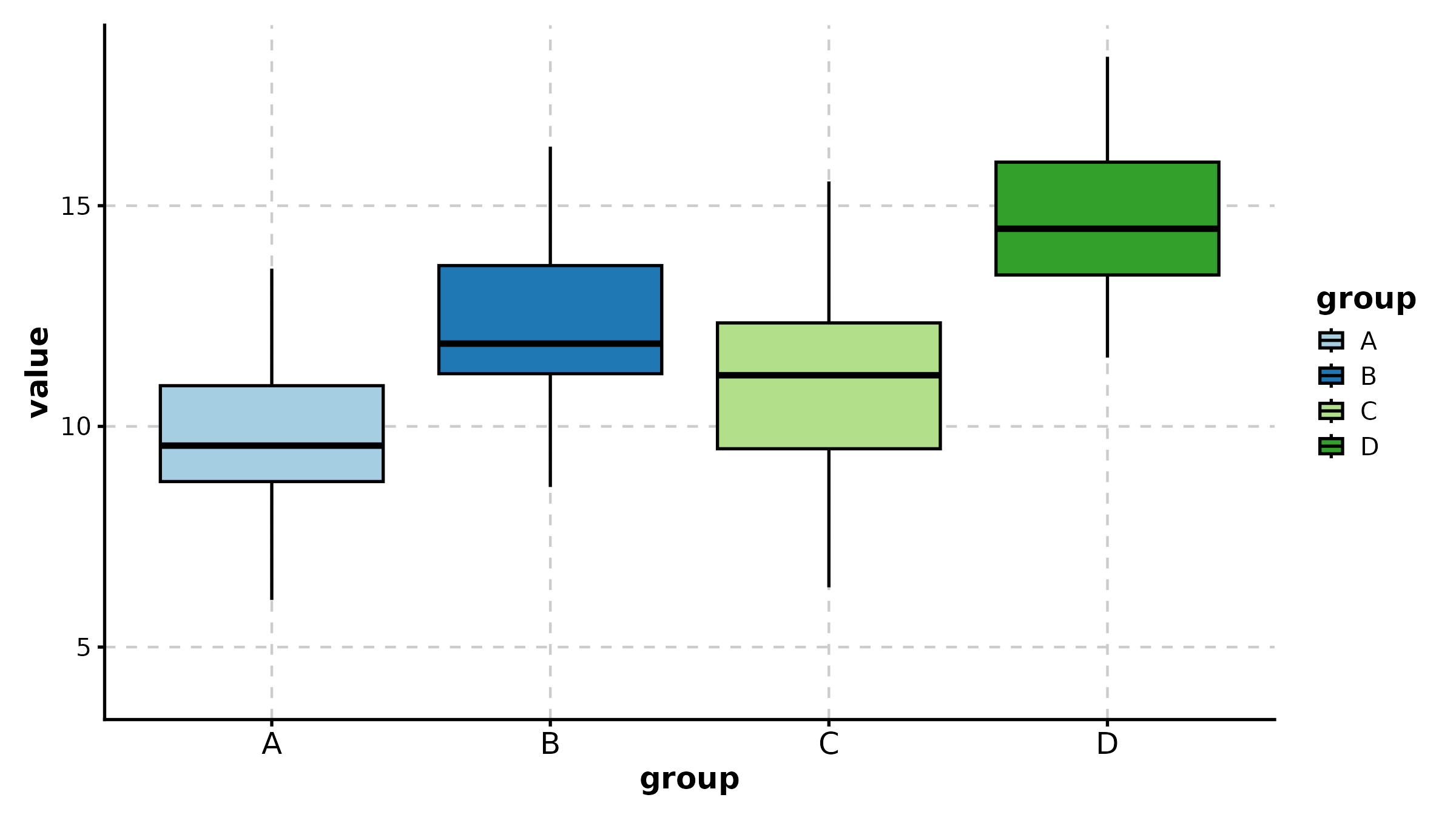
BoxPlot(treat, x = "group", y = "response", group_by = "sex",
palette = "npg", add_point = TRUE, pt_alpha = 0.4,
comparisons = TRUE, title = "Grouped with Comparisons")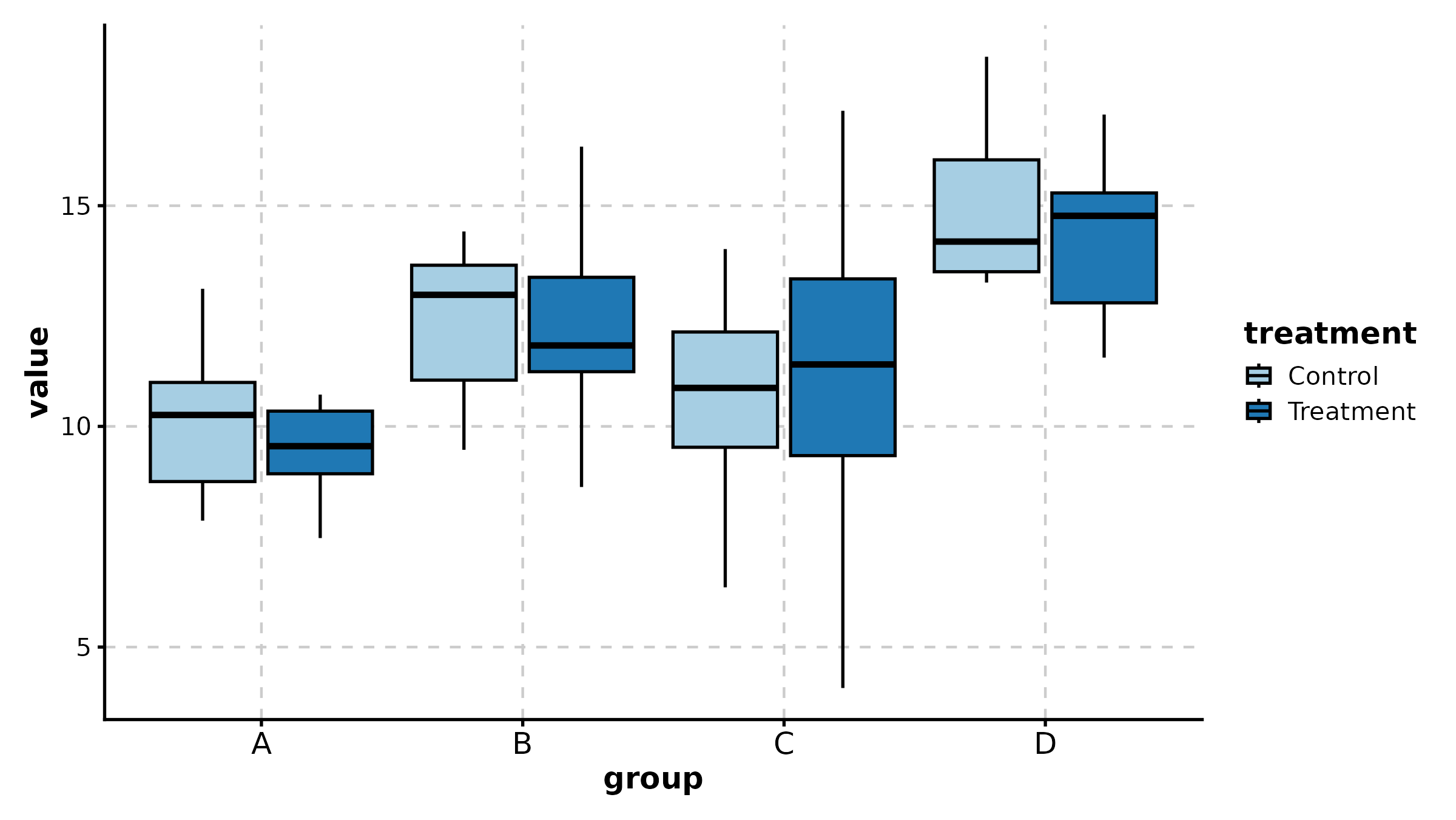
BoxPlot(treat, x = "group", y = "response", palette = "Set2",
fill_mode = "mean", comparisons = list(c("Control", "High Dose")),
sig_label = "p.signif", title = "Fill by Mean + Significance Symbols")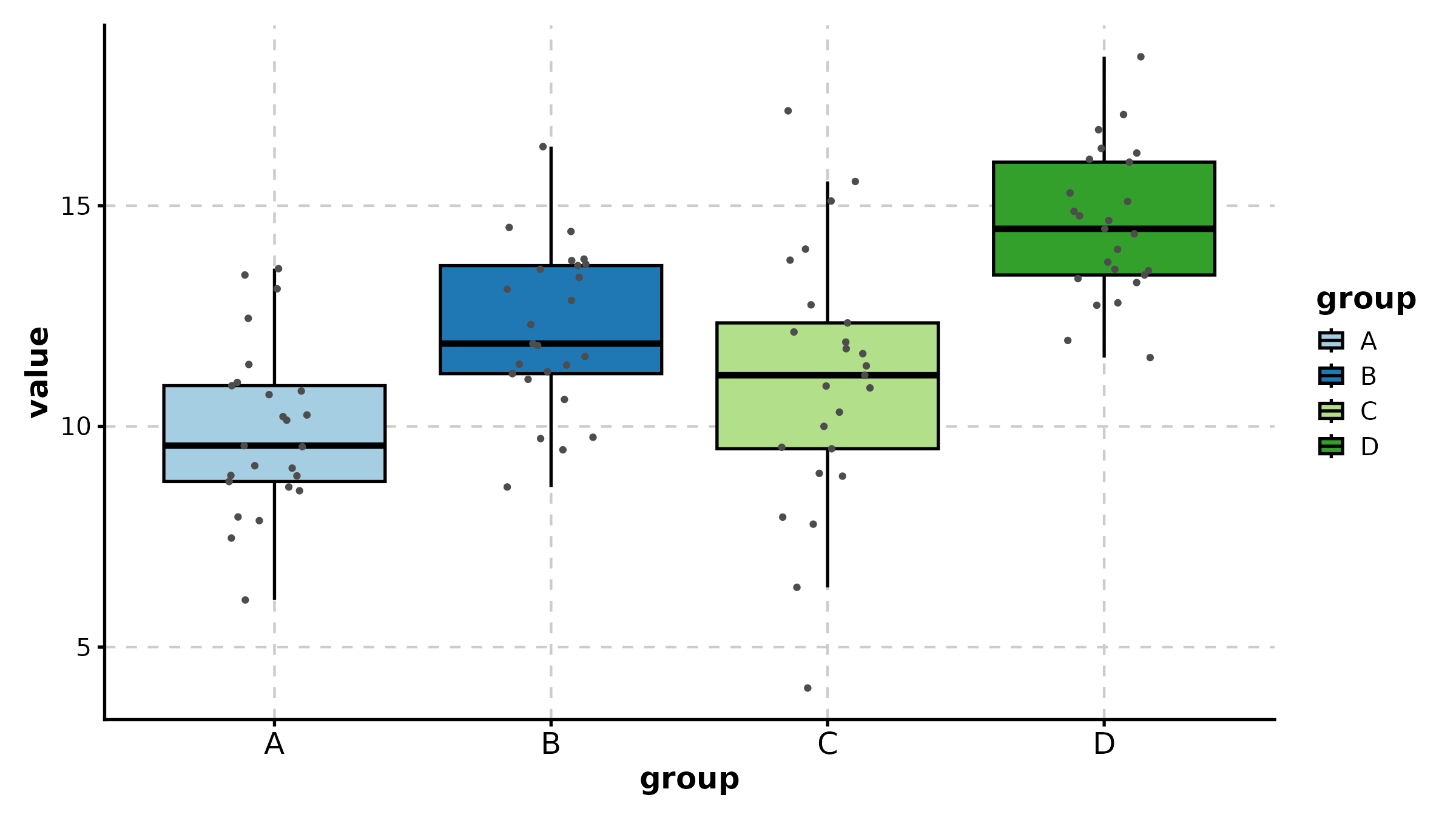
ViolinPlot
ViolinPlot(treat, x = "group", y = "response", palette = "npg",
add_box = TRUE, add_point = TRUE, pt_alpha = 0.3,
title = "Violin with Box Overlay")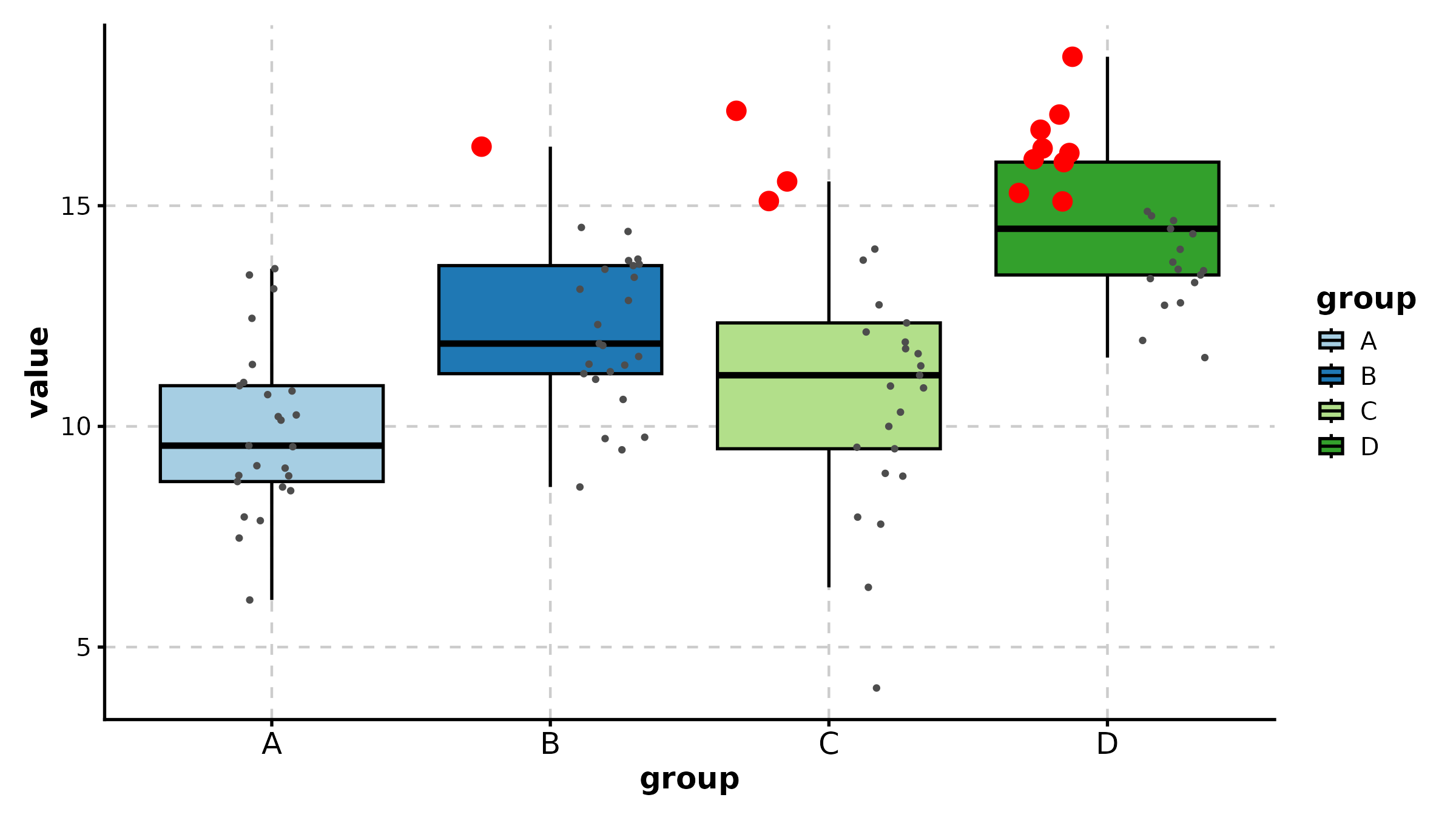
ViolinPlot(treat, x = "group", y = "response", group_by = "sex",
palette = "jco", add_box = TRUE,
title = "Grouped Violin")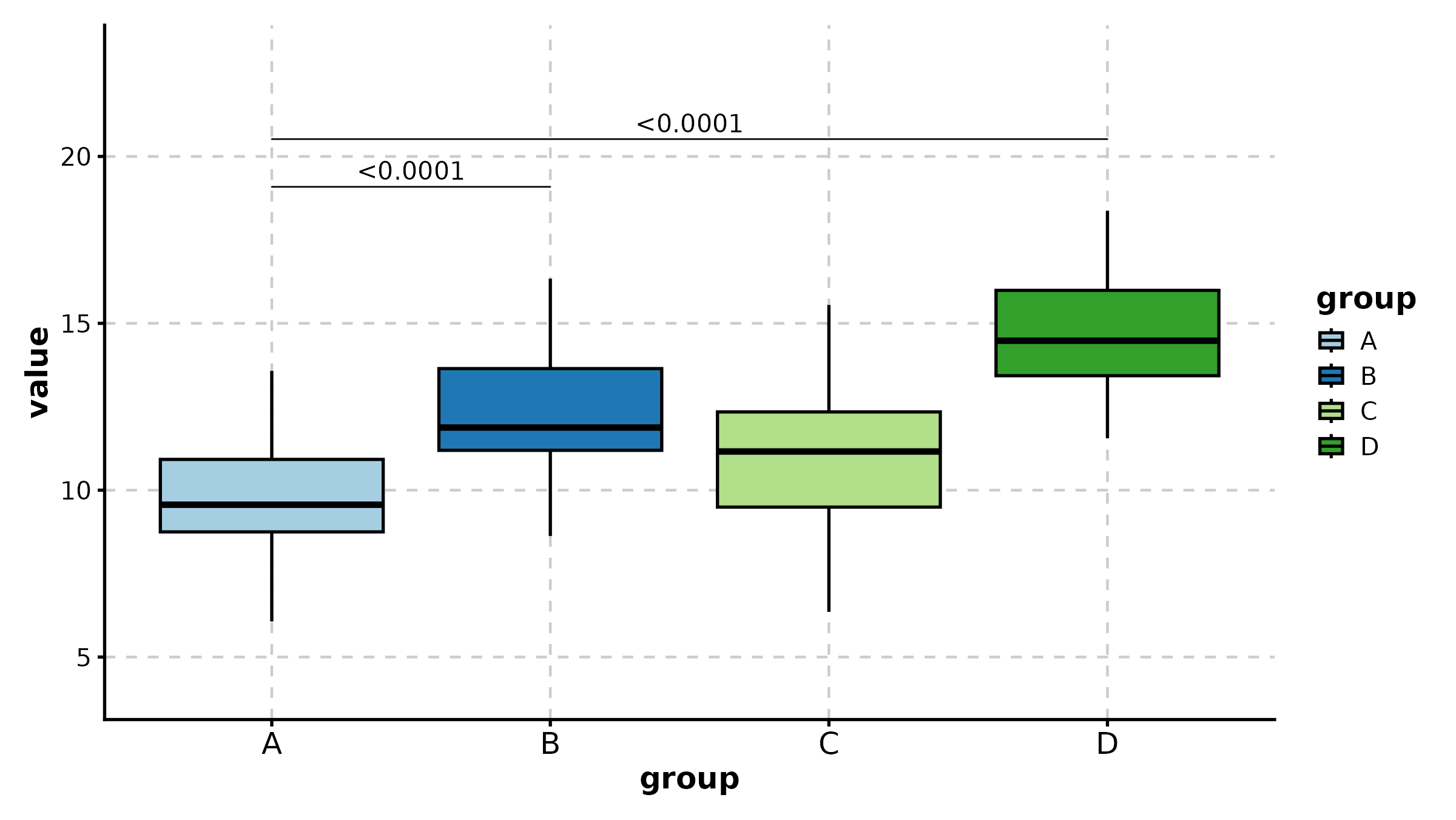
ViolinPlot(treat, x = "group", y = "response",
fill_mode = "median", palette = "RdYlBu",
title = "Fill by Median (Gradient)")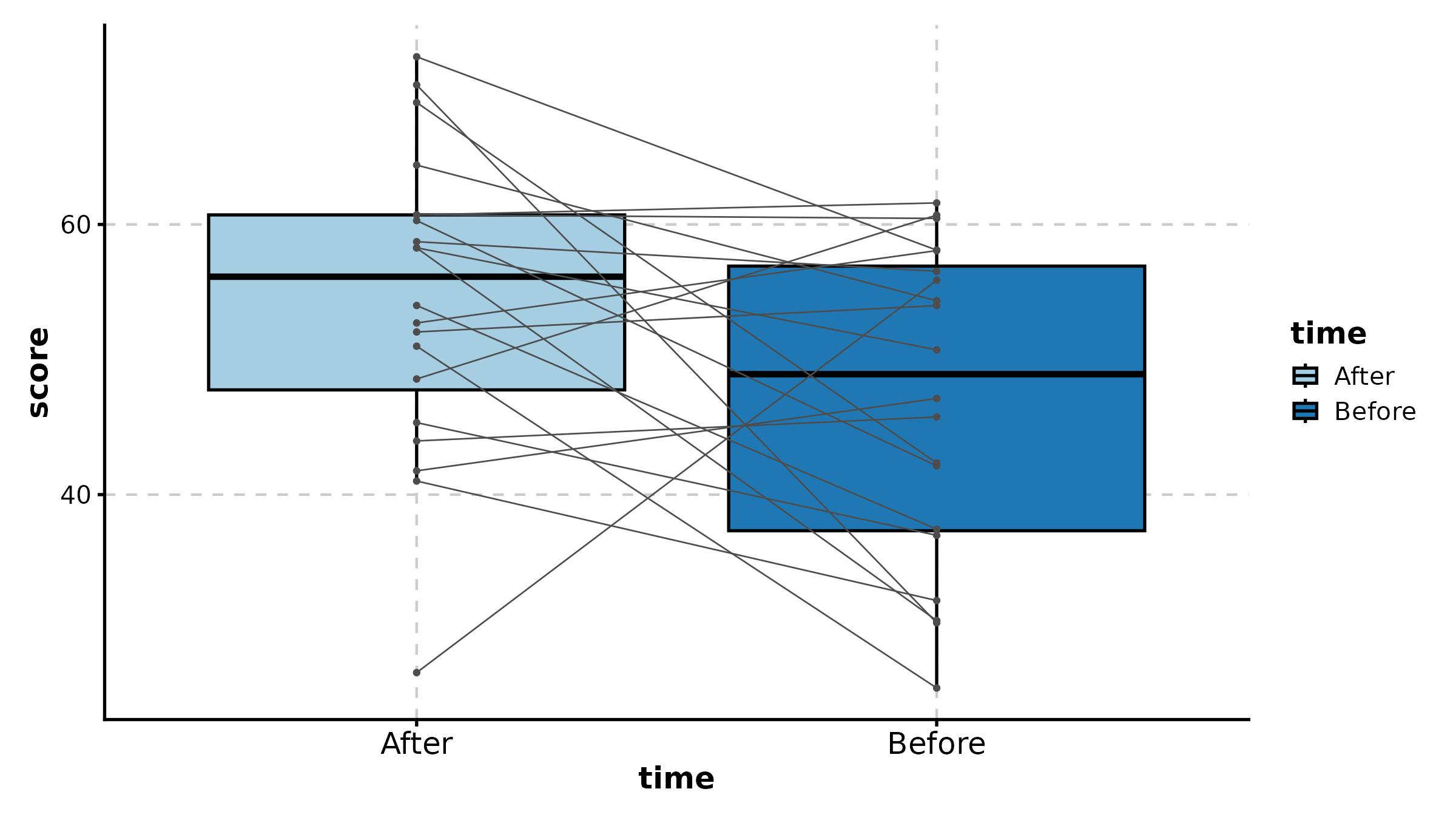
ScatterPlot
sdata <- data.frame(
expr_A = rnorm(120, 8, 2), expr_B = rnorm(120, 8, 2),
tissue = sample(c("Normal", "Tumor"), 120, replace = TRUE)
)
sdata$expr_B <- sdata$expr_B + 0.6 * sdata$expr_A + rnorm(120, 0, 1.5)
ScatterPlot(sdata, x = "expr_A", y = "expr_B", group_by = "tissue",
palette = "jco", add_smooth = TRUE, add_stat = TRUE,
title = "Grouped Correlation")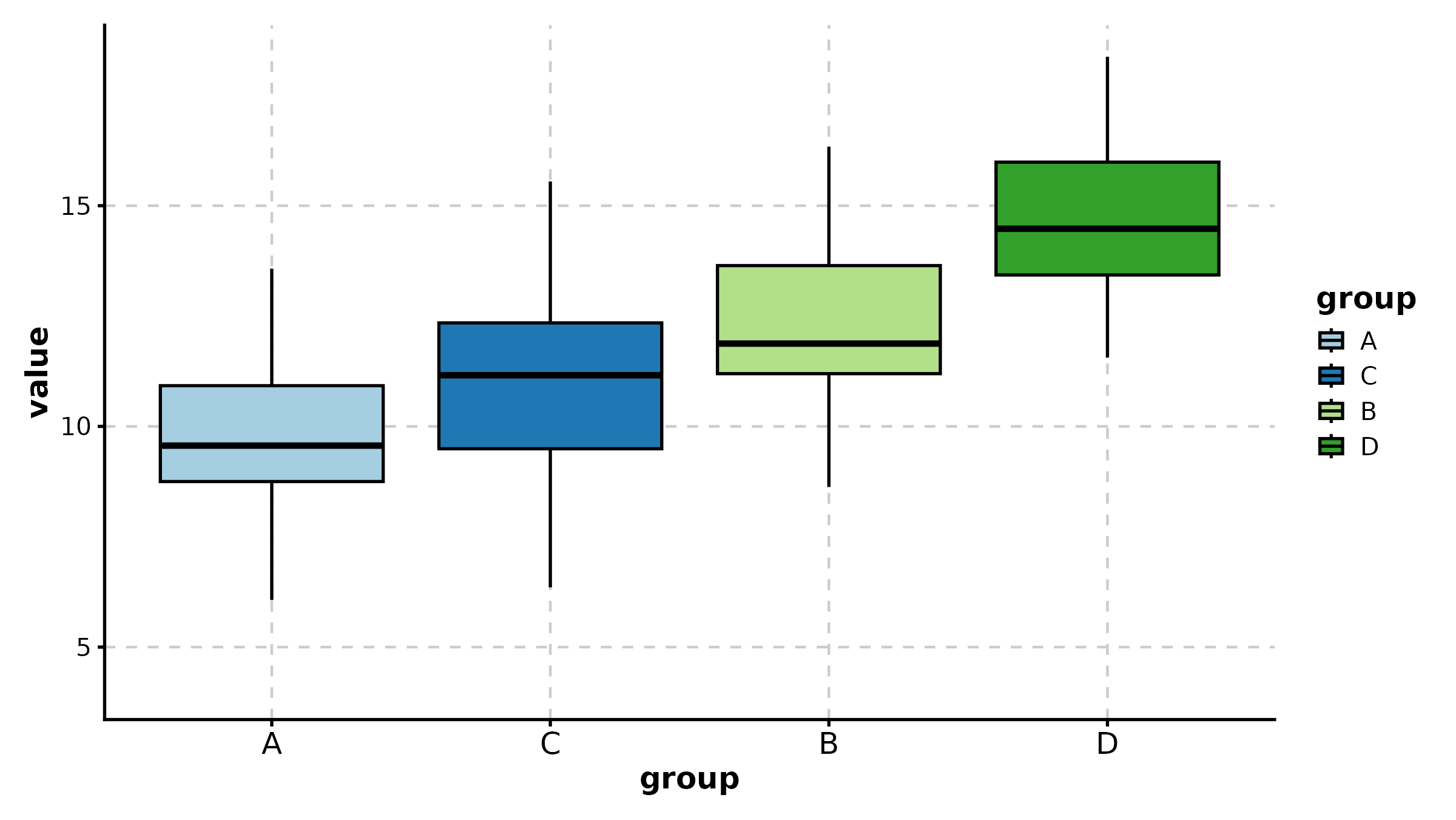
ScatterPlot(sdata, x = "expr_A", y = "expr_B",
color_by = "expr_B", palette = "viridis",
title = "Continuous Color Mapping")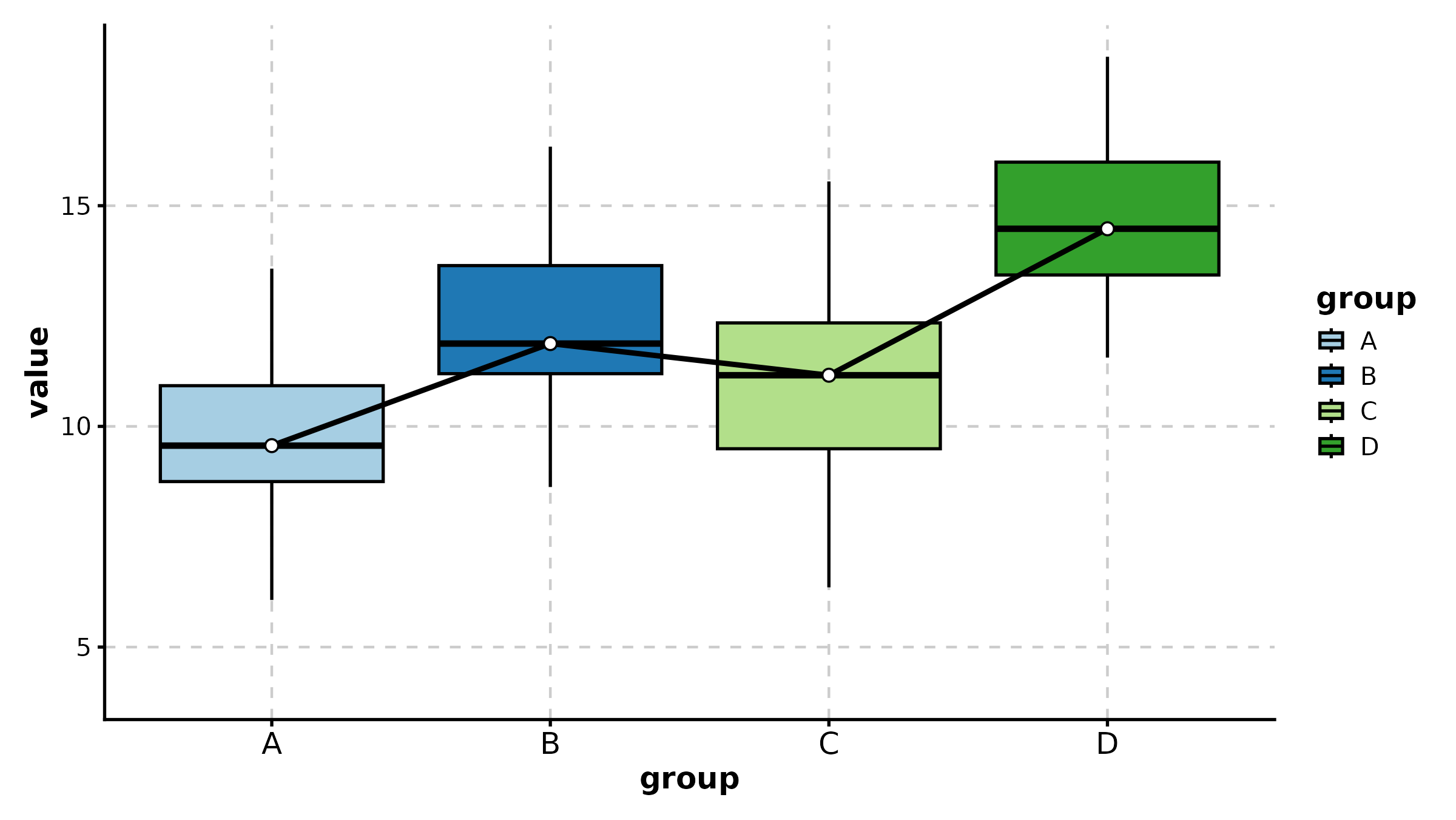
ScatterPlot(sdata, x = "expr_A", y = "expr_B",
group_by = "tissue", split_by = "tissue",
palette = "npg", add_smooth = TRUE,
combine = TRUE, nrow = 1,
title = "Split by Tissue")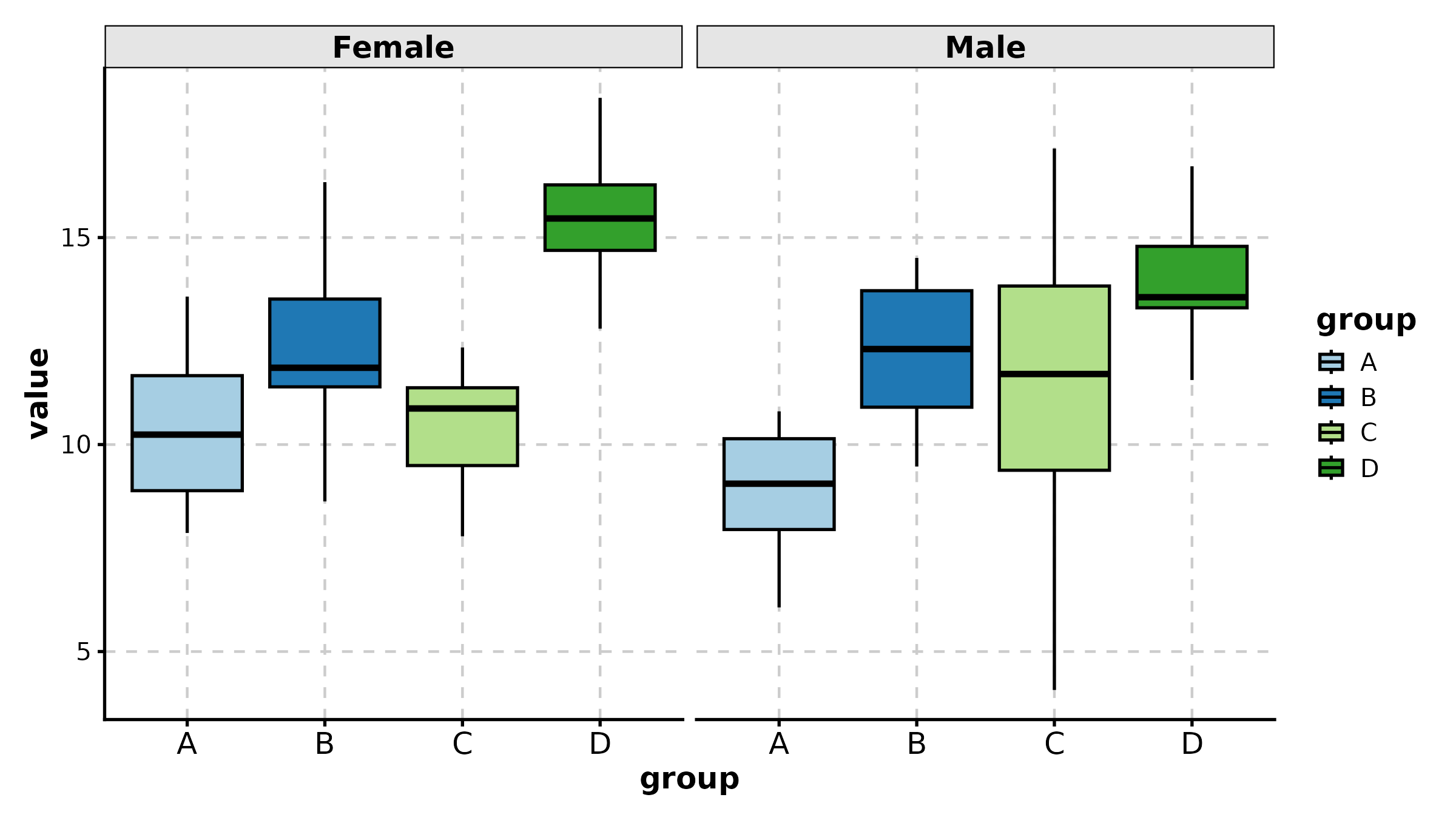
LinePlot
time_course <- data.frame(
day = rep(c(0, 3, 7, 14, 21, 28), 2),
volume = c(100, 120, 180, 320, 500, 780, 100, 110, 130, 150, 170, 200),
arm = rep(c("Vehicle", "Treatment"), each = 6)
)
LinePlot(time_course, x = "day", y = "volume", group_by = "arm",
palette = "lancet", title = "Tumor Growth Curve",
xlab = "Day", ylab = "Volume (mm3)")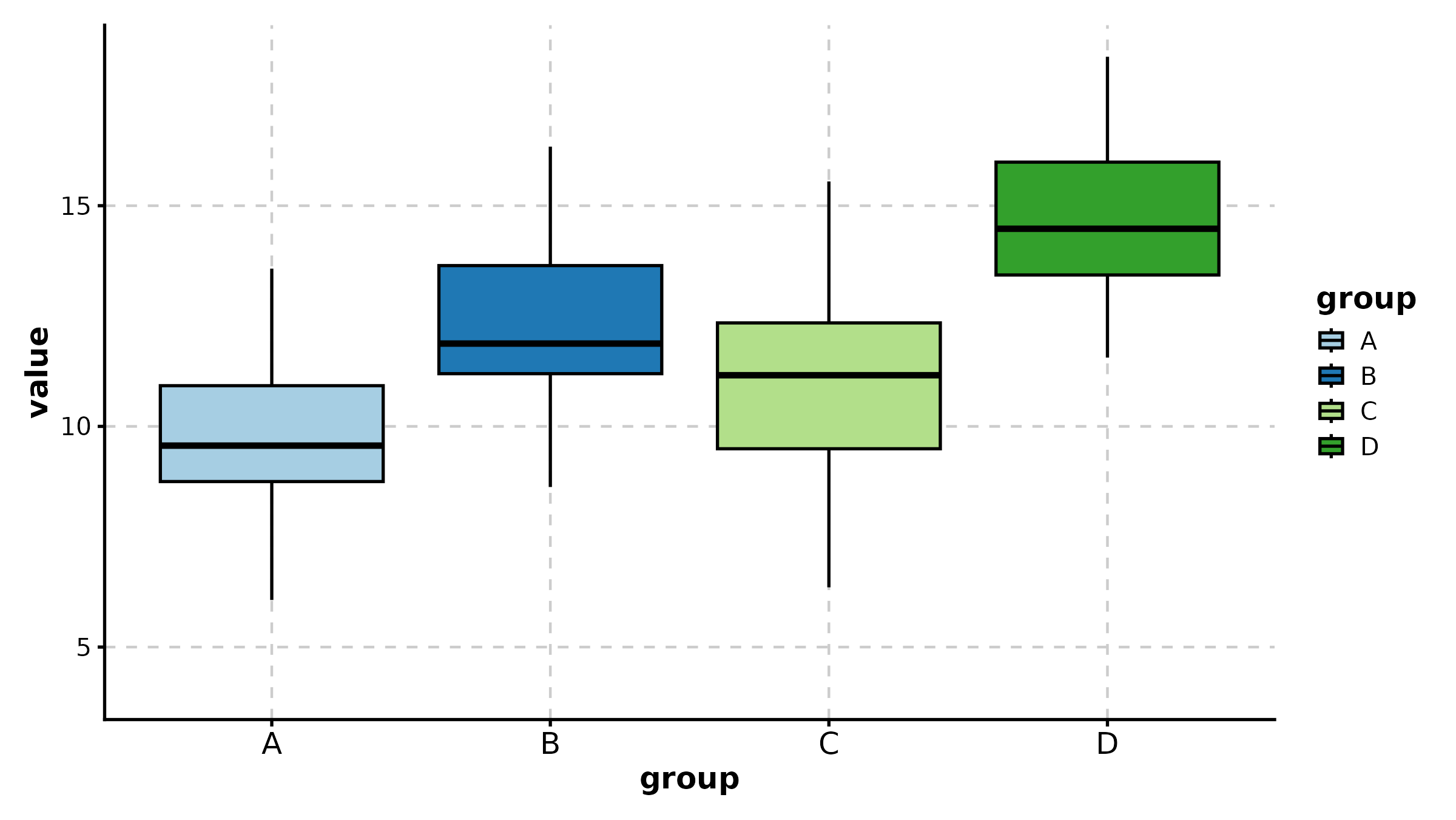
LinePlot(time_course, x = "day", y = "volume", group_by = "arm",
palette = "nejm", add_bg = TRUE, add_smooth = TRUE,
title = "With Background and Smooth")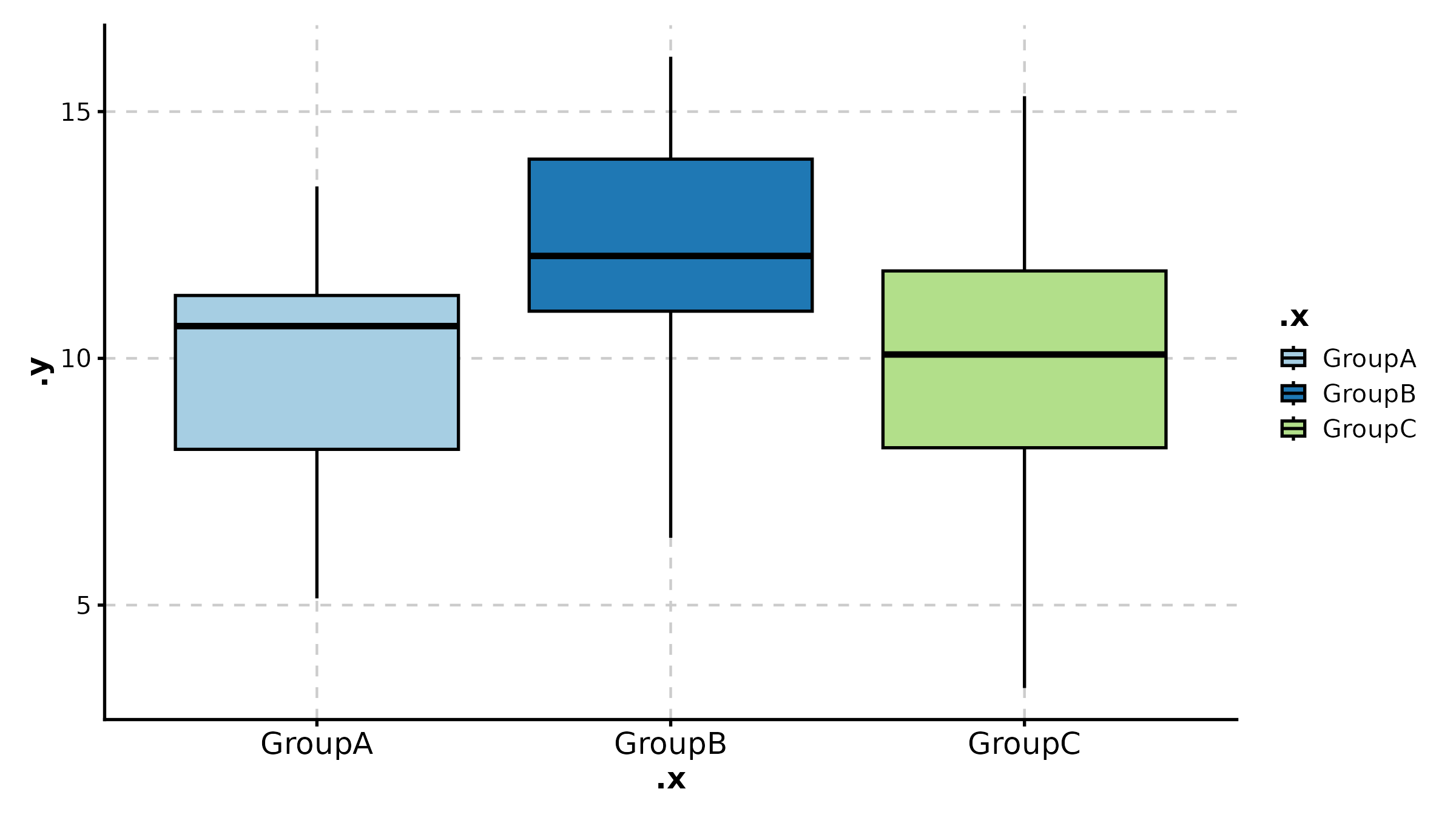
BarPlot
cell_freq <- data.frame(
sample = rep(c("Patient 1", "Patient 2", "Patient 3"), each = 4),
celltype = rep(c("T cell", "B cell", "Myeloid", "Stromal"), 3),
fraction = c(0.45, 0.15, 0.25, 0.15, 0.30, 0.25, 0.30, 0.15,
0.50, 0.10, 0.20, 0.20)
)
BarPlot(cell_freq, x = "sample", y = "fraction", group_by = "celltype",
position = "stack", palette = "npg", label = TRUE,
title = "Stacked Bar Plot")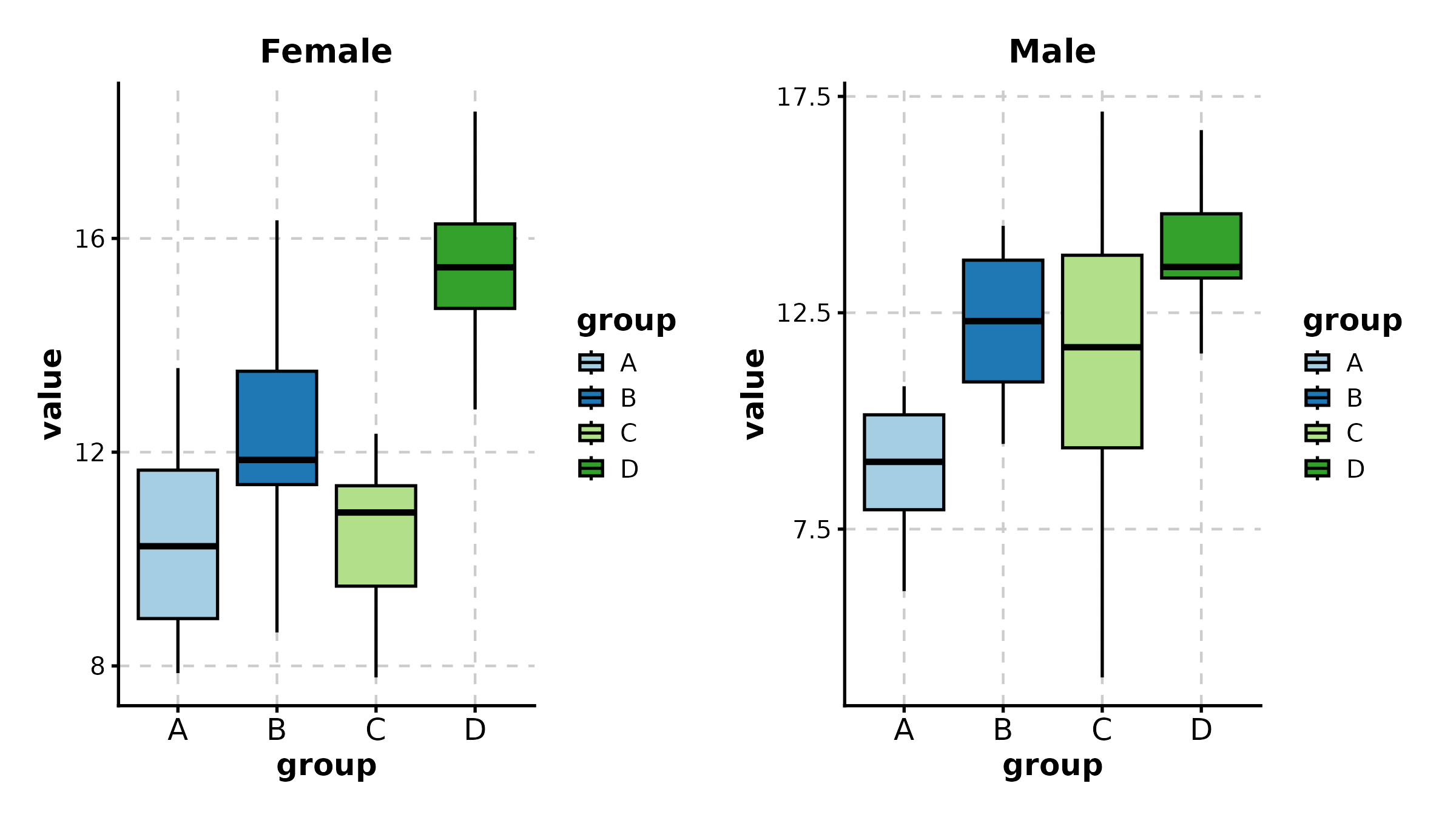
BarPlot(cell_freq, x = "sample", y = "fraction", group_by = "celltype",
position = "dodge", palette = "Set2",
title = "Grouped (Dodged) Bar Plot")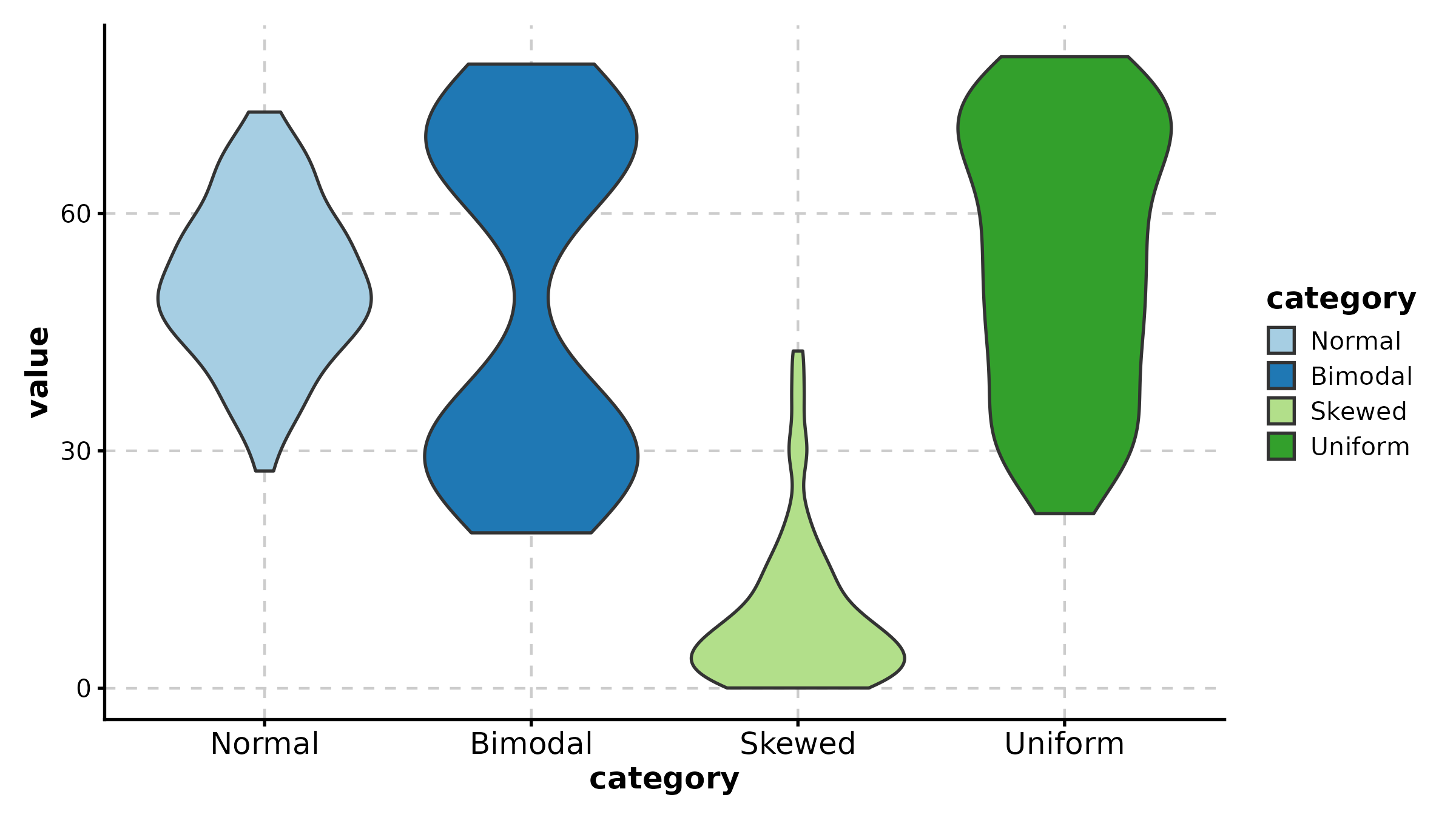
BarPlot(treat, x = "group", y = "response", palette = "lancet",
add_errorbar = TRUE, errorbar_type = "se",
title = "Mean with Standard Error")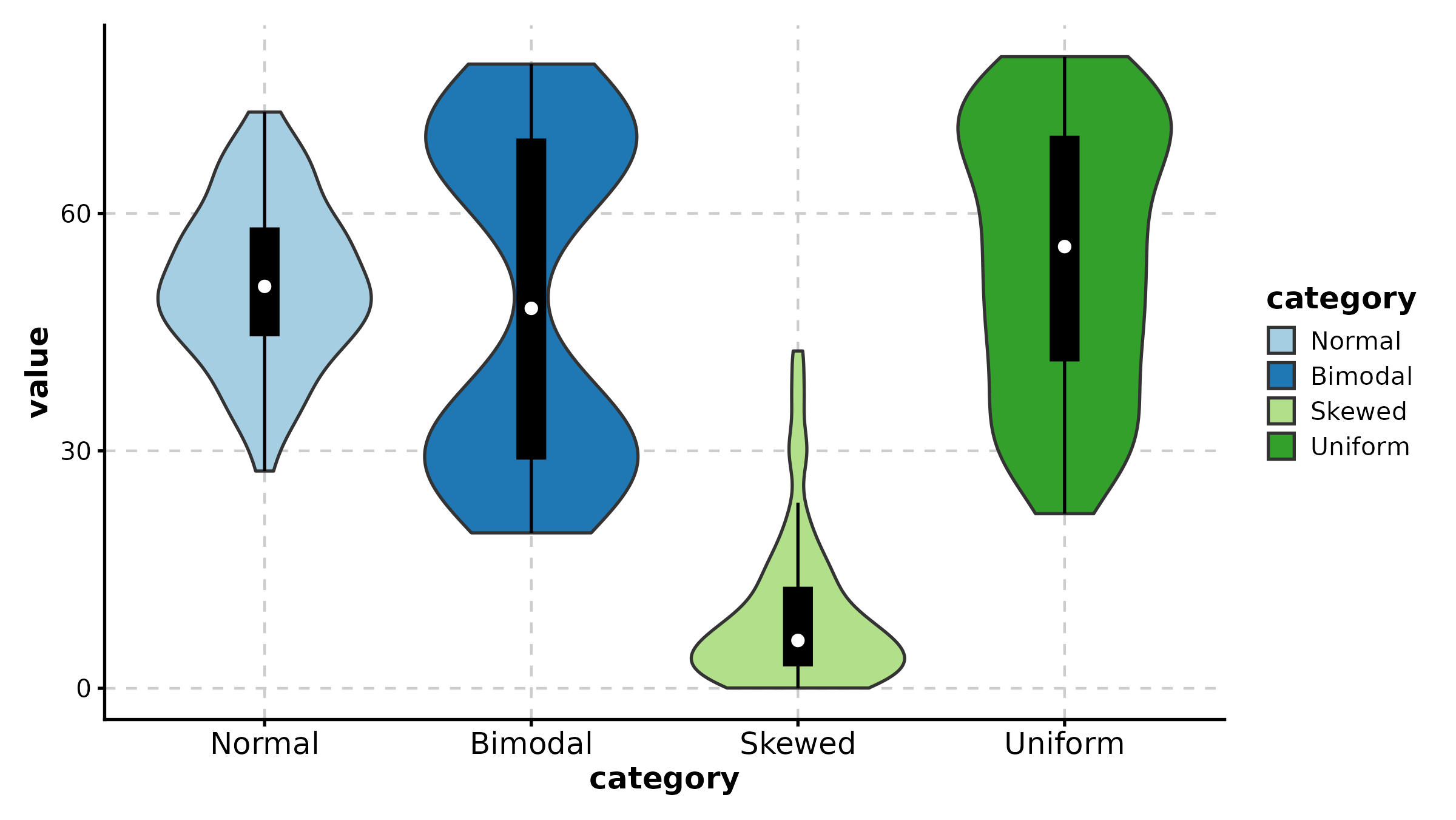
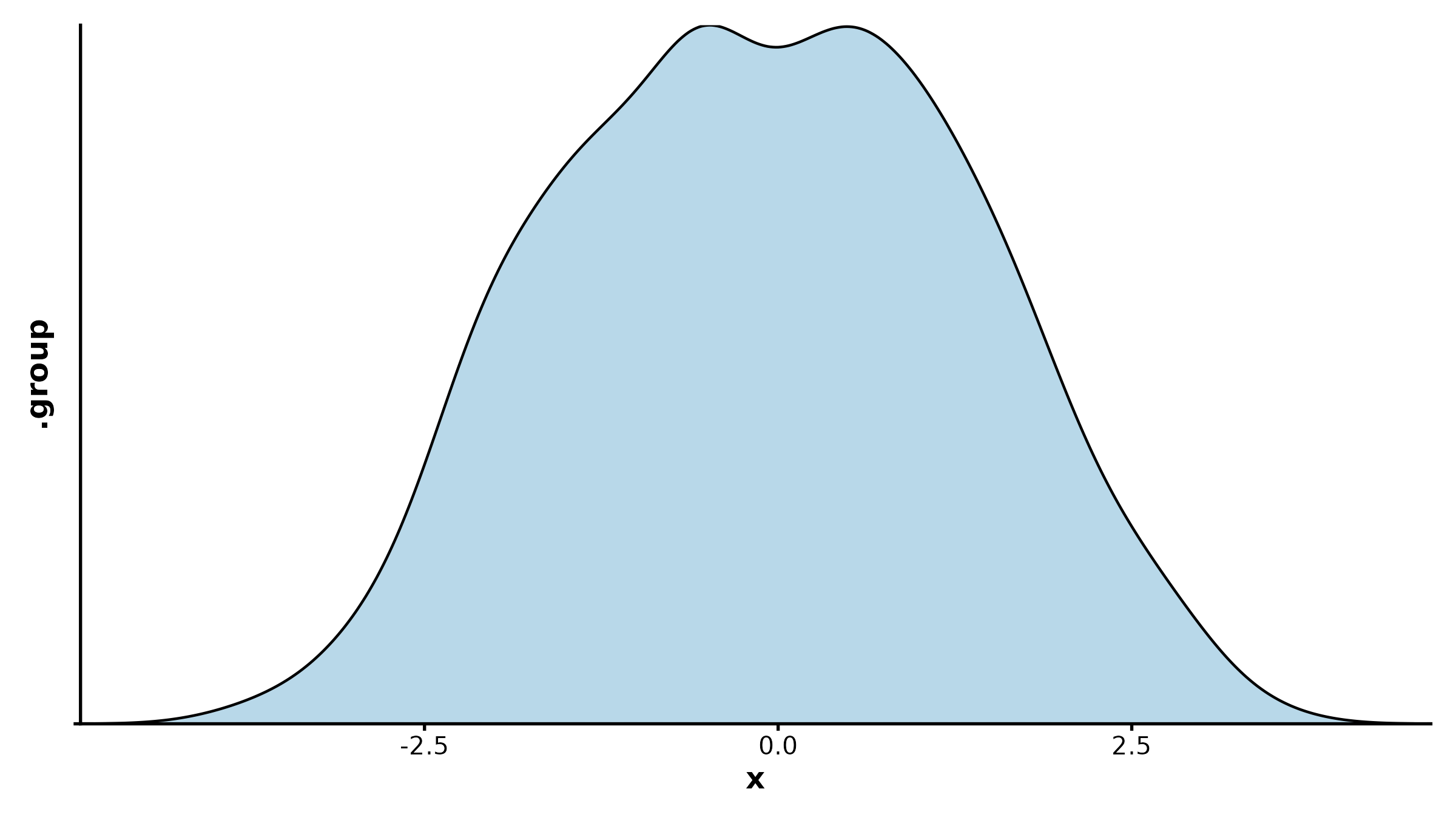
DensityPlot
DensityPlot(treat, x = "response", group_by = "group",
palette = "npg", add_rug = TRUE,
title = "Density with Rug")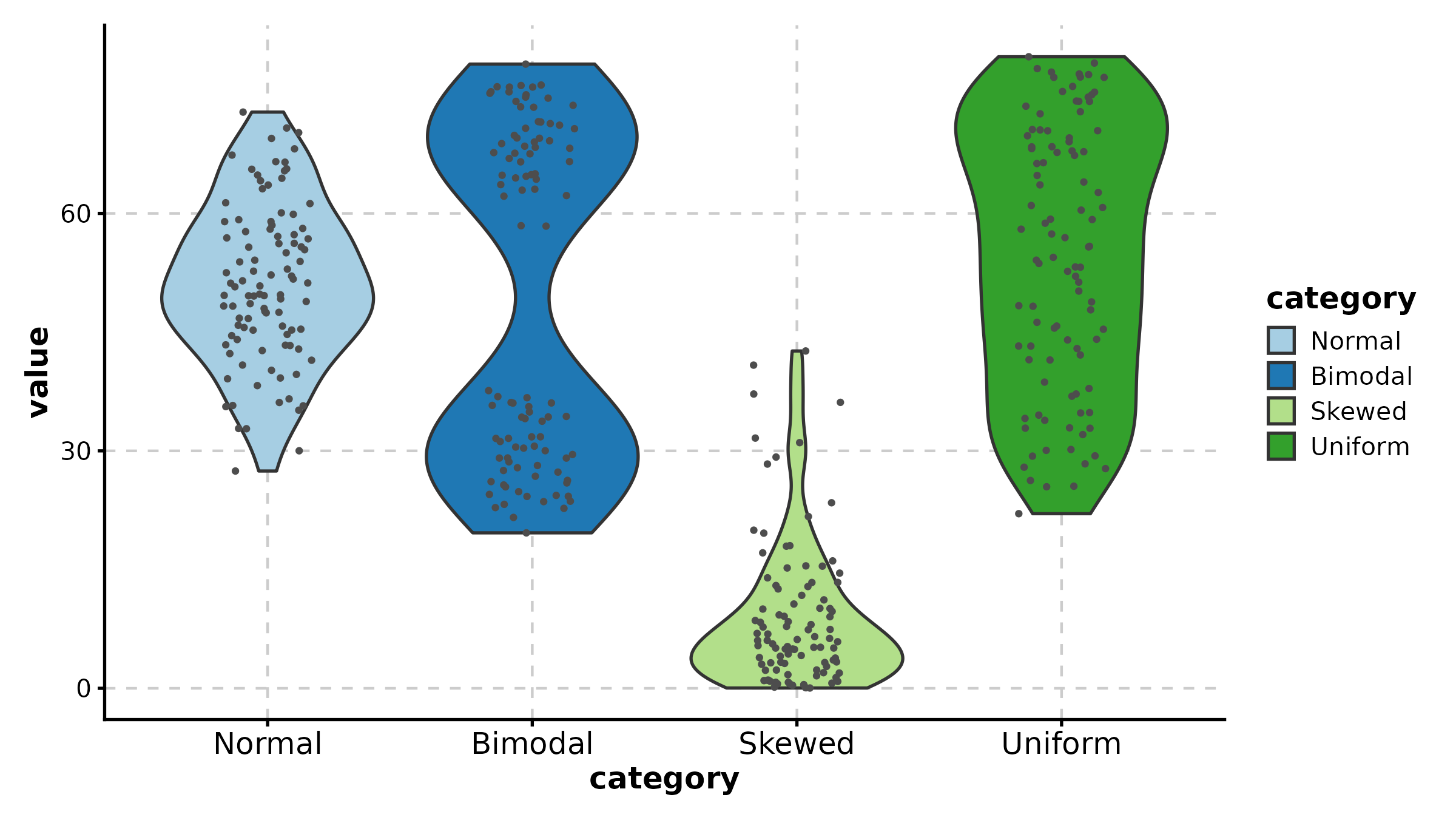
DensityPlot(treat, x = "response", group_by = "group",
facet_by = "sex", palette = "Set1",
title = "Faceted Density")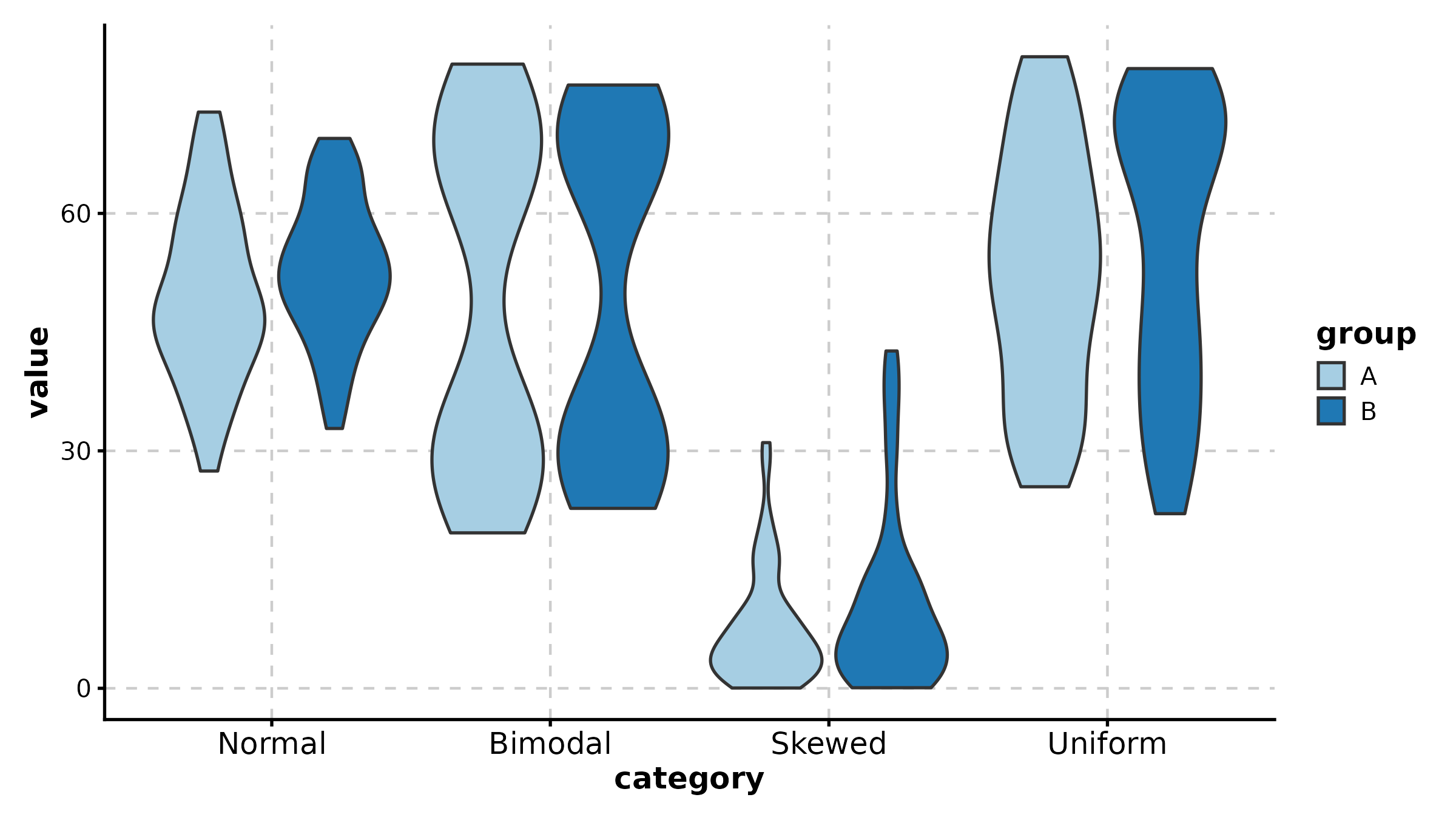
JitterPlot
JitterPlot(treat, x = "group", y = "response", palette = "Set2",
title = "Basic Jitter Plot")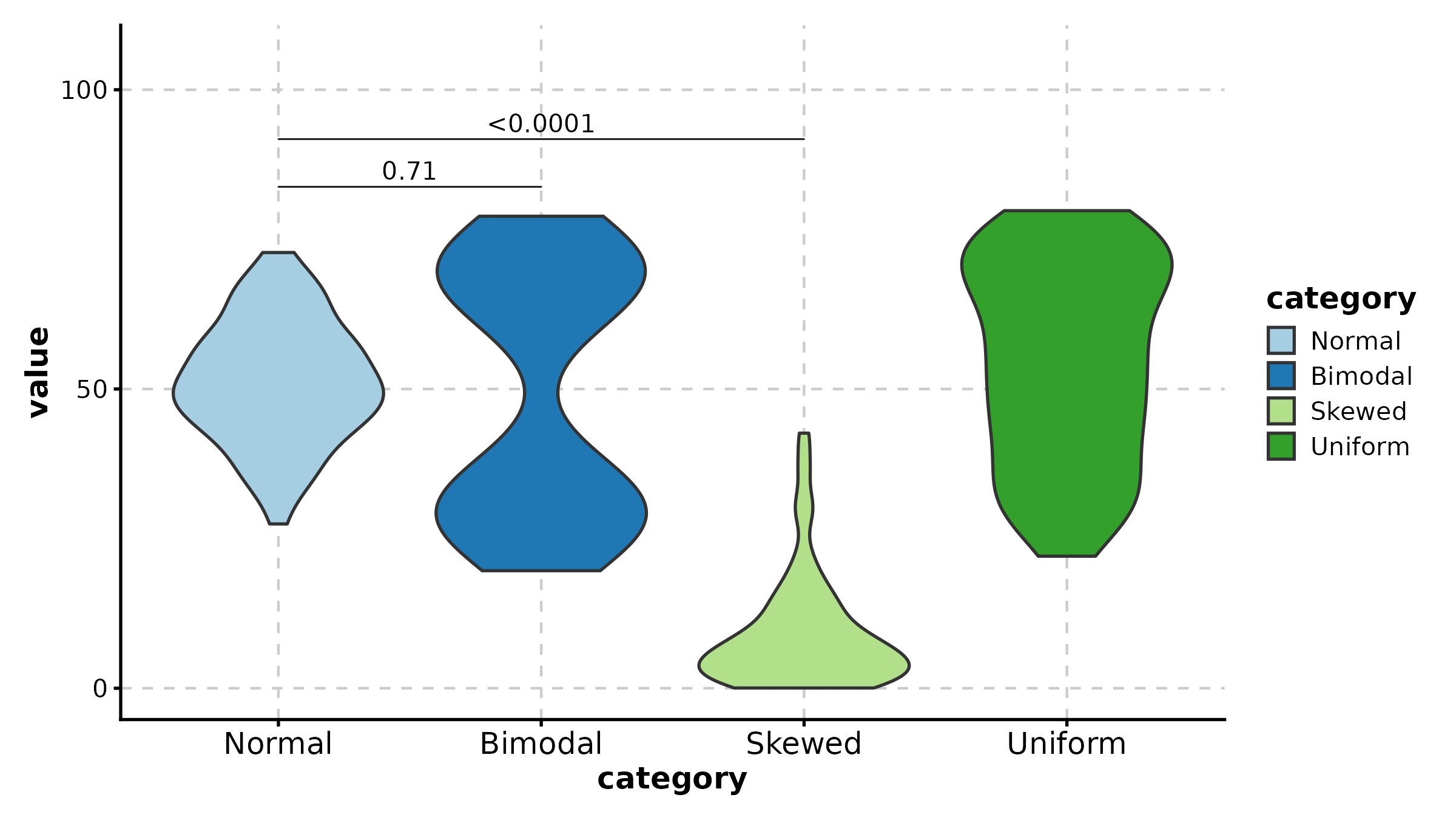
JitterPlot(treat, x = "group", y = "response", group_by = "sex",
palette = "npg", title = "Grouped Jitter")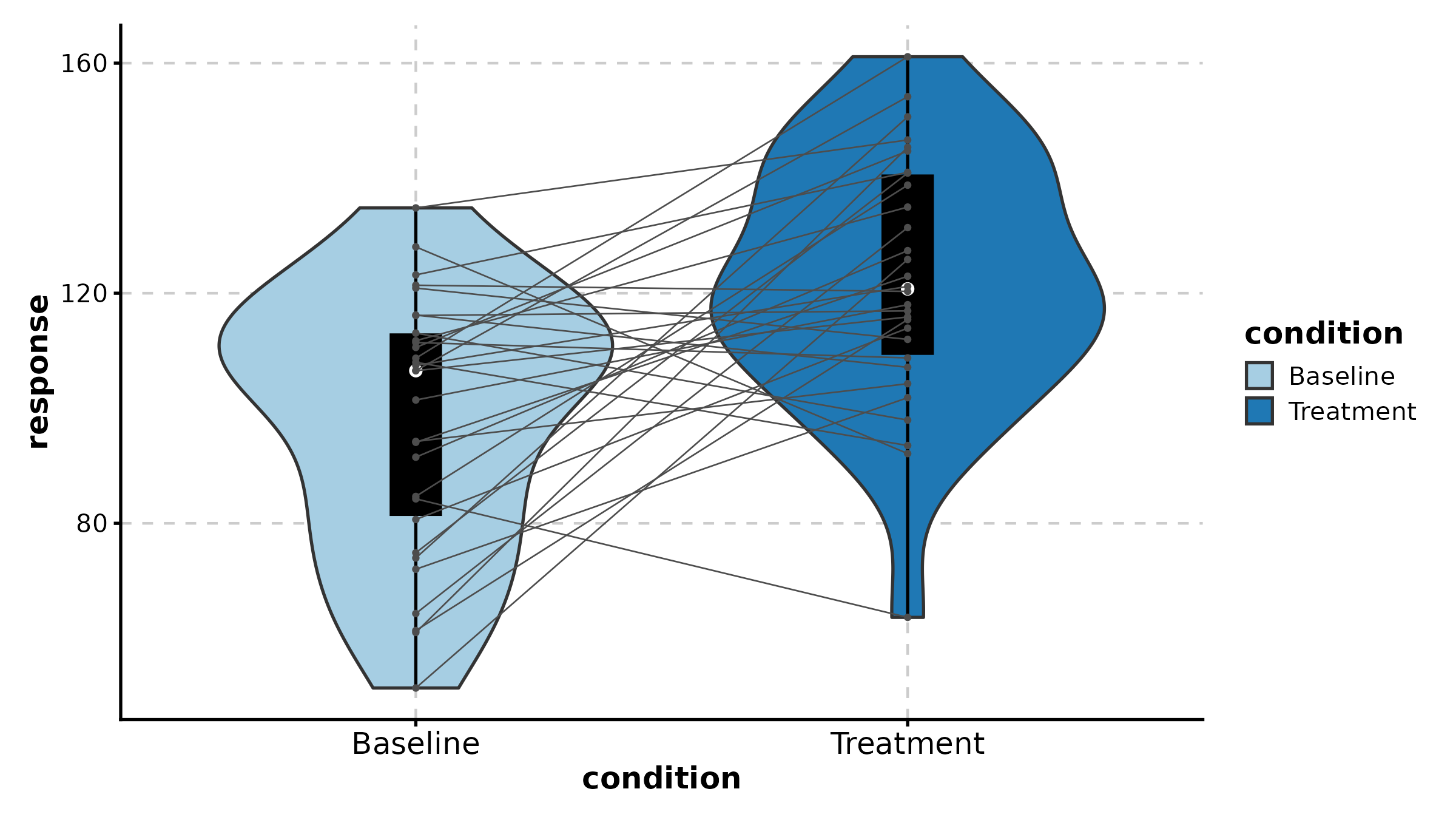
BeeswarmPlot
BeeswarmPlot(treat, x = "group", y = "response", palette = "npg",
title = "Beeswarm Layout")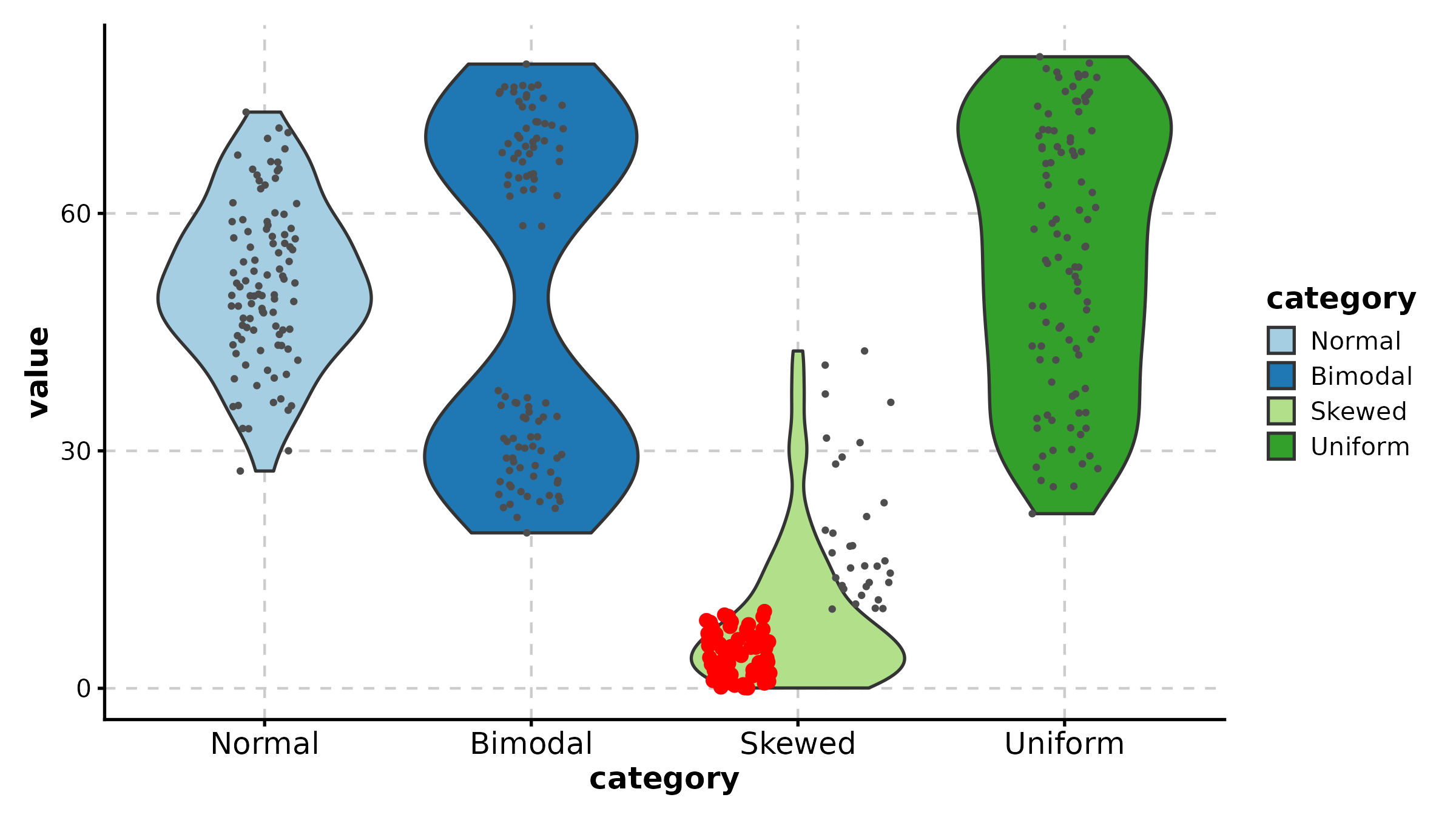
Histogram
Histogram(data.frame(score = c(rnorm(200, 5, 2), rnorm(200, 10, 2)),
group = rep(c("A", "B"), each = 200)),
x = "score", group_by = "group", palette = "Set1",
title = "Histogram")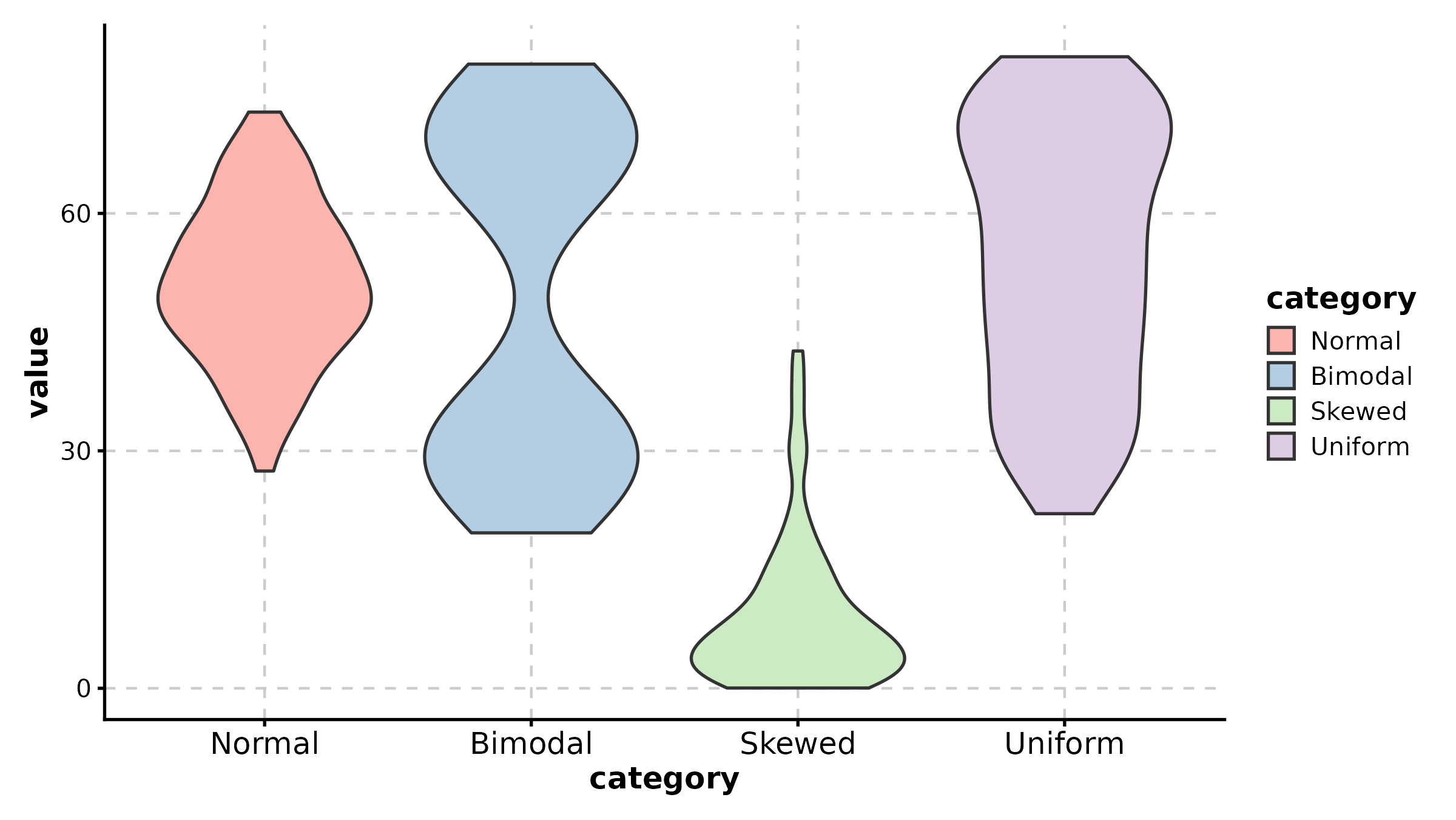
RidgePlot
ridge_df <- data.frame(
value = c(rnorm(100, 3), rnorm(100, 5), rnorm(100, 7), rnorm(100, 9)),
celltype = rep(c("T cell", "B cell", "Myeloid", "NK"), each = 100)
)
RidgePlot(ridge_df, x = "value", group_by = "celltype",
palette = "npg", title = "Ridge Plot")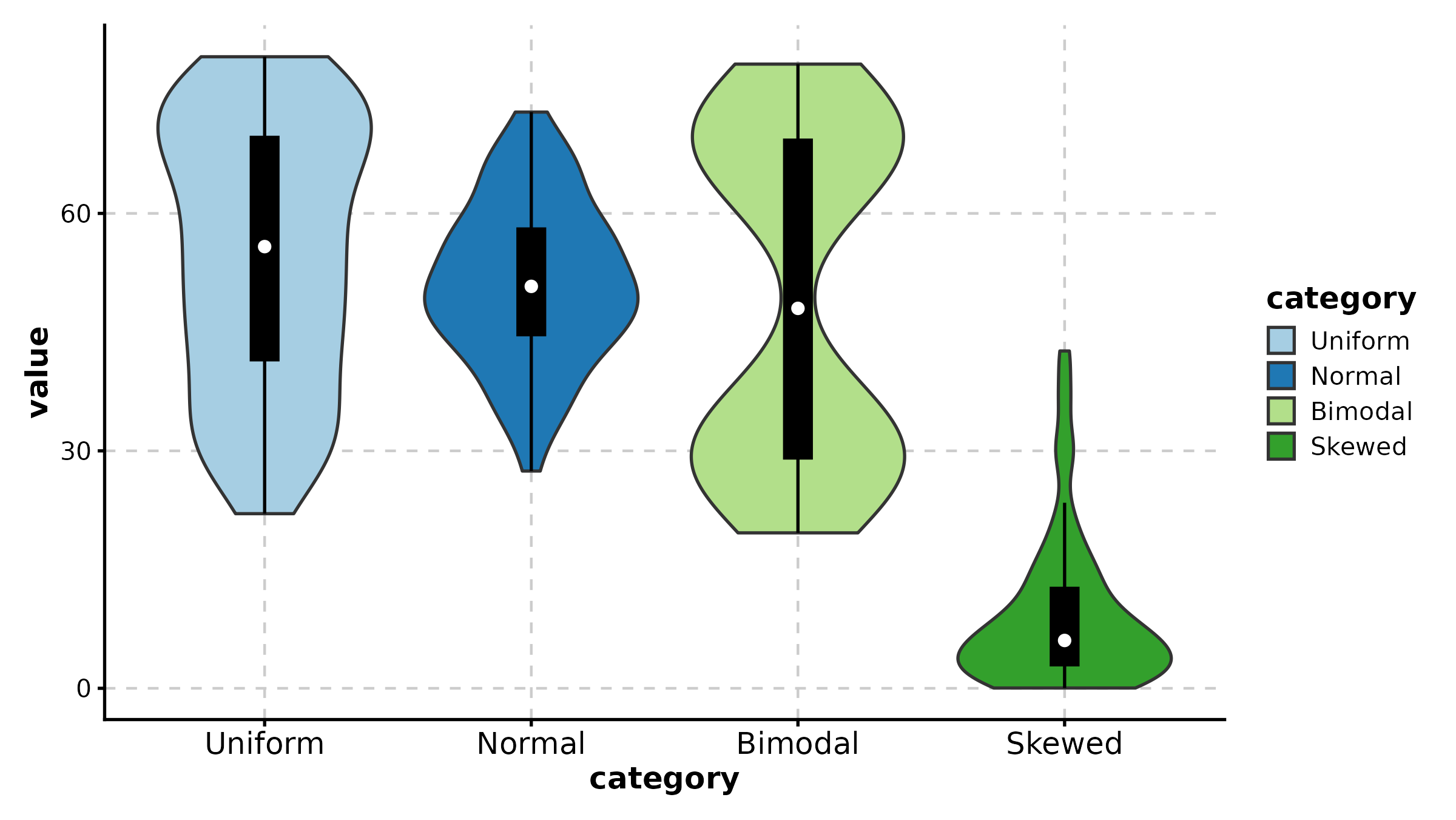
RidgePlot(ridge_df, x = "value", group_by = "celltype",
palette = "Set2", add_vline = TRUE, vline_color = TRUE,
title = "Ridge with Median Lines")
AreaPlot
comp <- data.frame(
time = rep(paste0("T", 1:6), 3),
fraction = c(0.50, 0.45, 0.40, 0.35, 0.30, 0.25,
0.30, 0.30, 0.35, 0.35, 0.40, 0.40,
0.20, 0.25, 0.25, 0.30, 0.30, 0.35),
celltype = rep(c("Epithelial", "Immune", "Stromal"), each = 6)
)
AreaPlot(comp, x = "time", y = "fraction", group_by = "celltype",
palette = "nejm", title = "Stacked Area")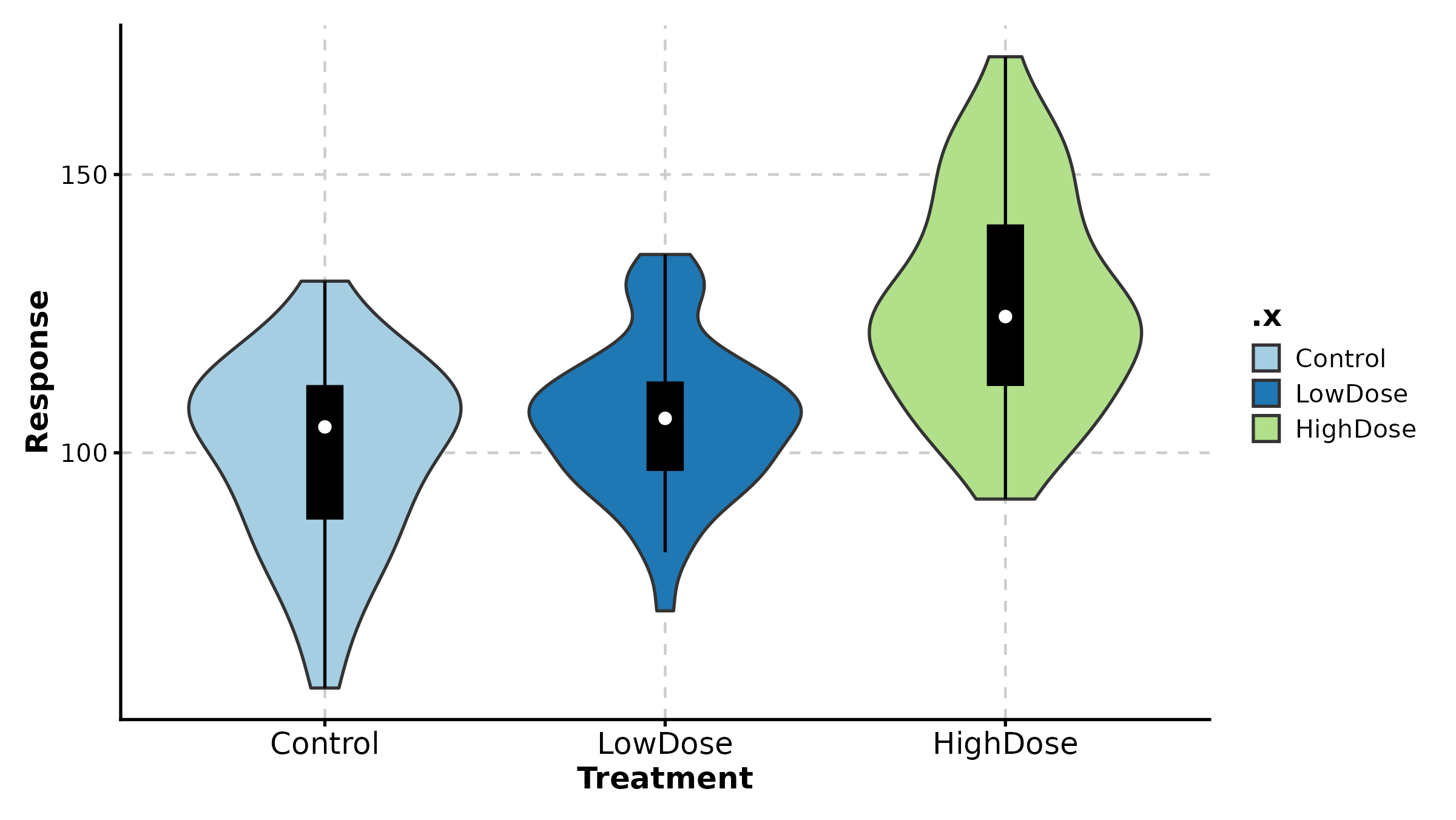
AreaPlot(comp, x = "time", y = "fraction", group_by = "celltype",
palette = "Set2", scale_y = TRUE,
title = "100% Stacked Area")
TrendPlot
trend_df <- data.frame(
timepoint = rep(LETTERS[1:5], 2),
value = c(2, 5, 8, 6, 3, 4, 7, 11, 9, 5),
cohort = rep(c("Cohort 1", "Cohort 2"), each = 5)
)
TrendPlot(trend_df, x = "timepoint", y = "value", group_by = "cohort",
palette = "Set1", title = "Trend Comparison")
TrendPlot(trend_df, x = "timepoint", y = "value", group_by = "cohort",
palette = "nejm", scale_y = TRUE,
title = "Scaled Trend")QQPlot
QQPlot(data.frame(values = rnorm(200)), val = "values",
band = TRUE, title = "Normal Q-Q Plot")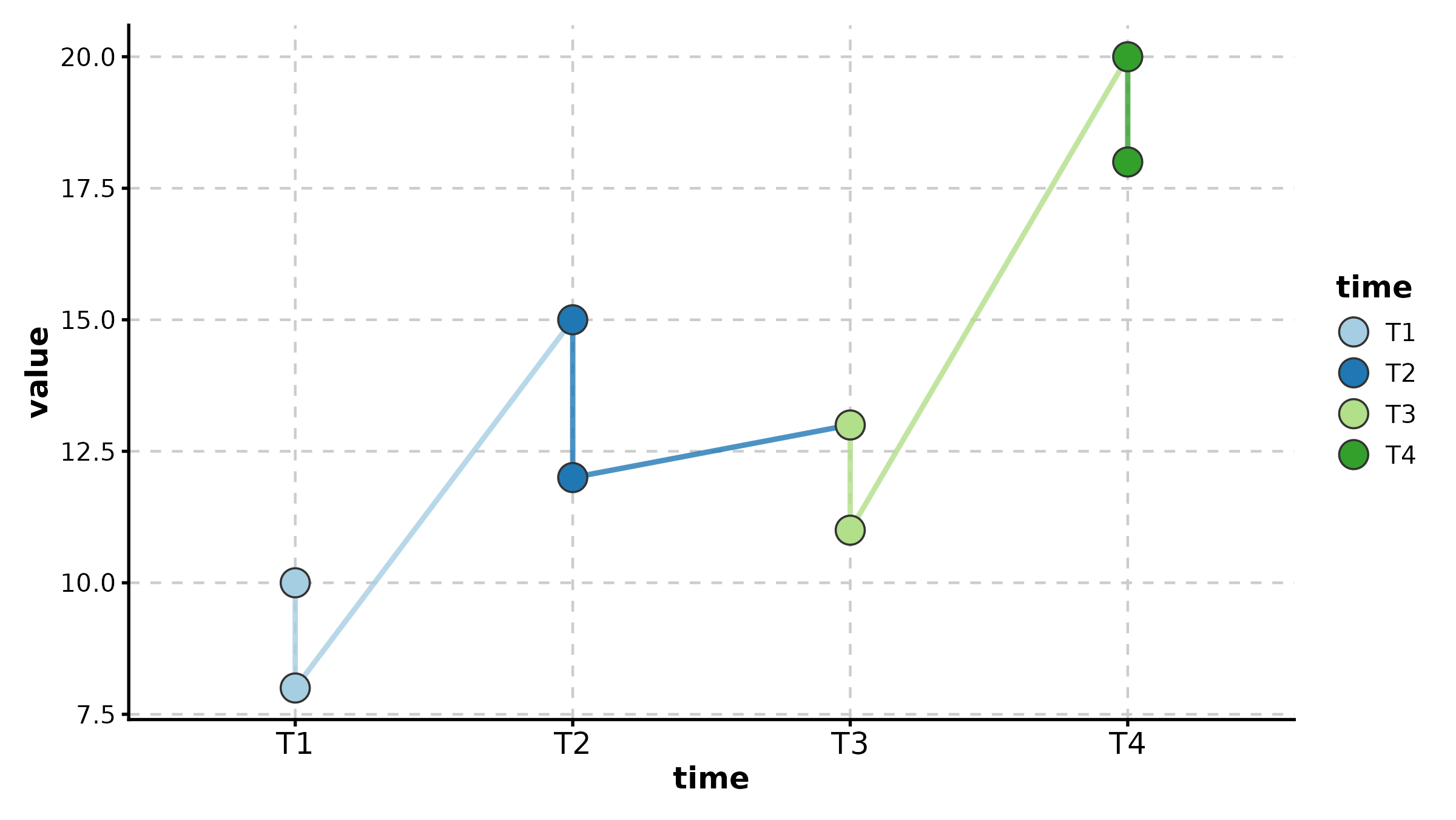
DotPlot
dot_df <- data.frame(
gene = rep(c("CD3D", "CD8A", "MS4A1", "CD68", "NKG7"), 4),
cluster = rep(paste0("C", 1:4), each = 5),
avg_expr = runif(20, -1, 2),
pct_expr = runif(20, 0.1, 0.9)
)
DotPlot(dot_df, x = "gene", y = "cluster",
size_by = "pct_expr", fill_by = "avg_expr",
palette = "RdBu", title = "Marker Expression Dot Plot")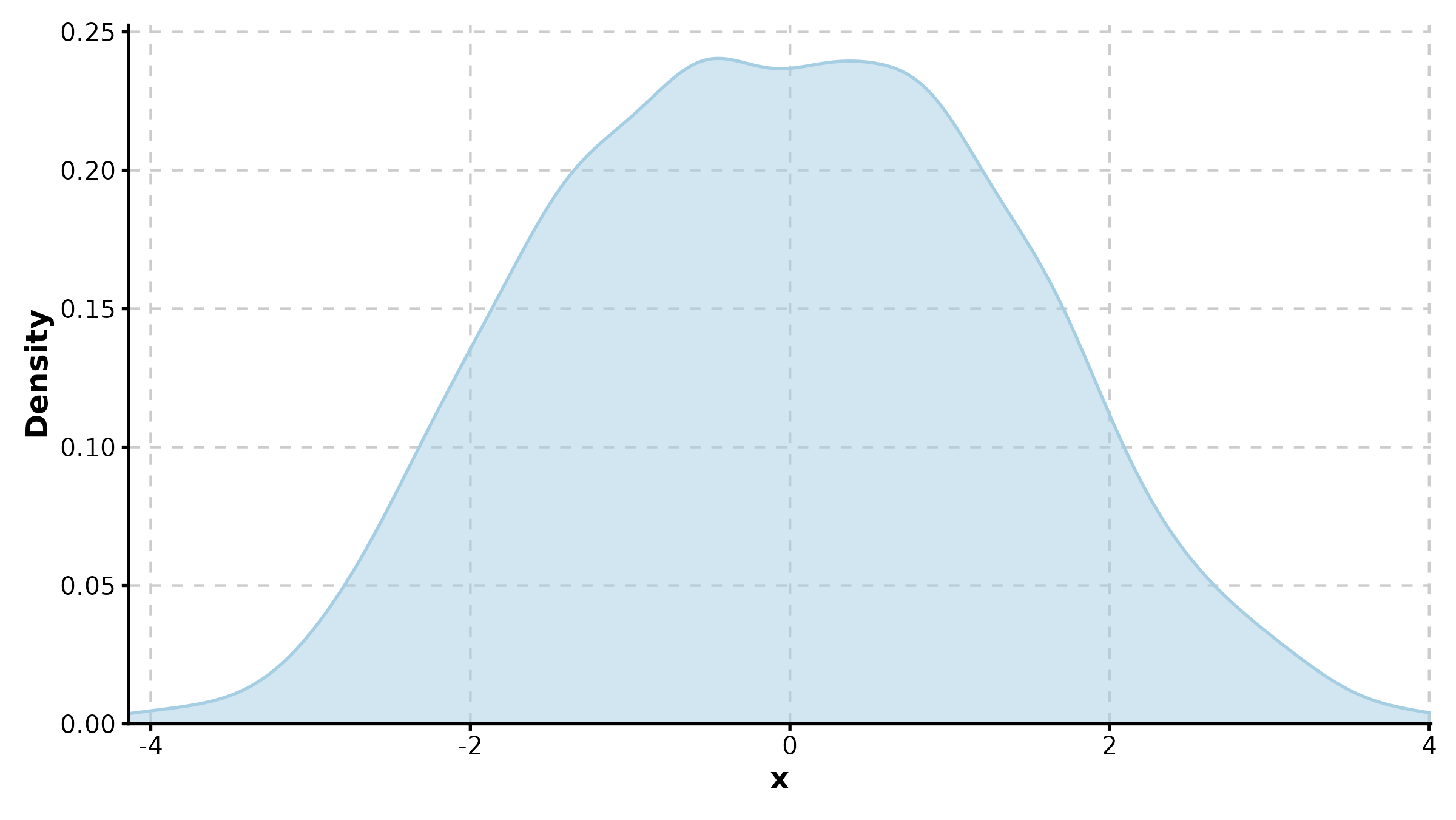
DotPlot(dot_df, x = "gene", y = "cluster",
size_by = "pct_expr", fill_by = "avg_expr",
fill_cutoff = 0, palette = "RdBu", add_bg = TRUE,
title = "With Background and Cutoff")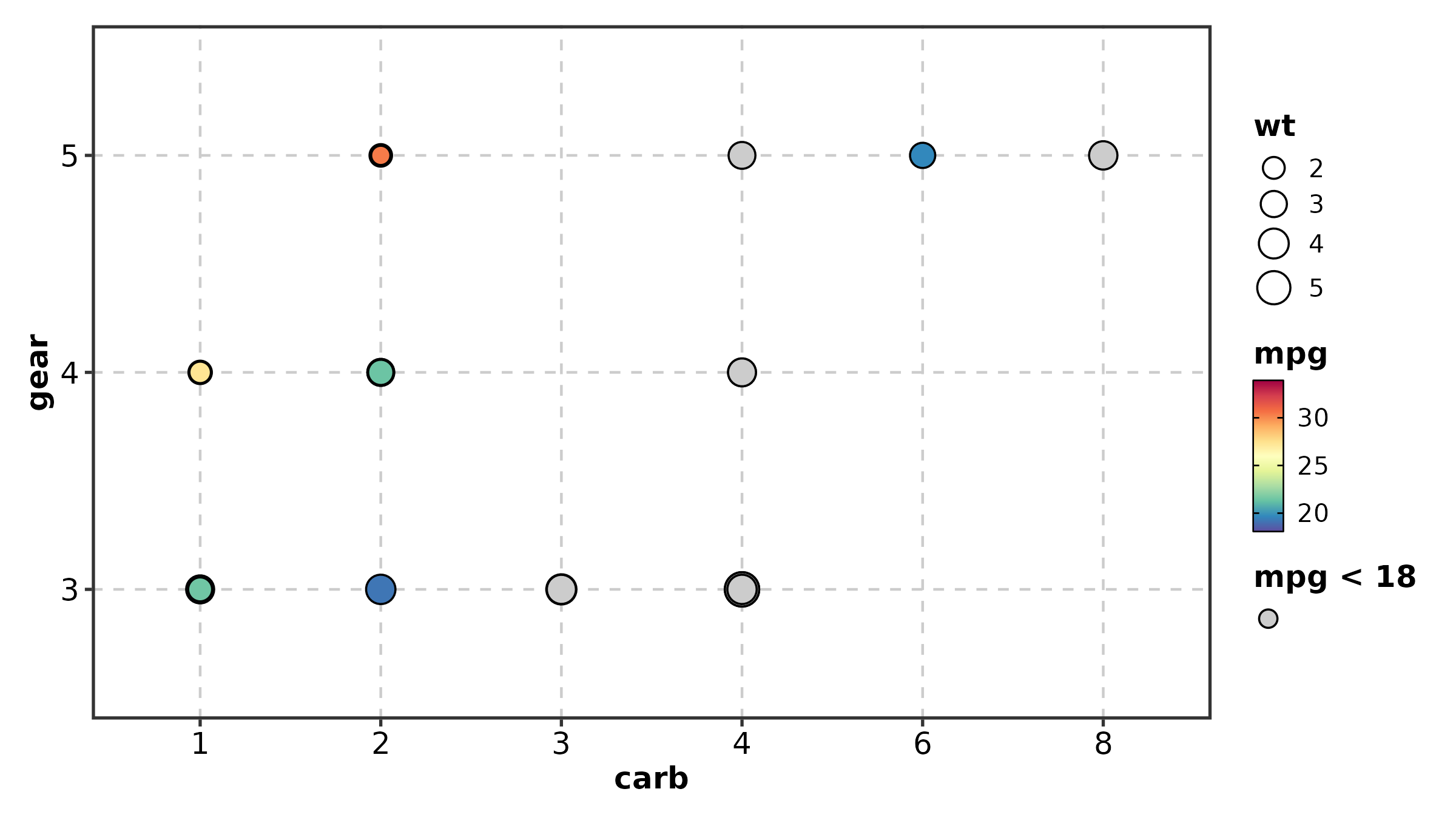
CorPlot
cor_df <- data.frame(
BRCA1 = rnorm(80), TP53 = rnorm(80),
tissue = sample(c("Normal", "Tumor"), 80, replace = TRUE)
)
cor_df$TP53 <- cor_df$BRCA1 * 0.7 + rnorm(80, sd = 0.5)
CorPlot(cor_df, x = "BRCA1", y = "TP53", group_by = "tissue",
palette = "jco", add_smooth = TRUE, title = "Gene Correlation")
CorPlot(cor_df, x = "BRCA1", y = "TP53",
palette = "lancet", add_smooth = TRUE,
anno_items = c("n", "eq", "r2", "pearson"),
title = "With Full Annotations")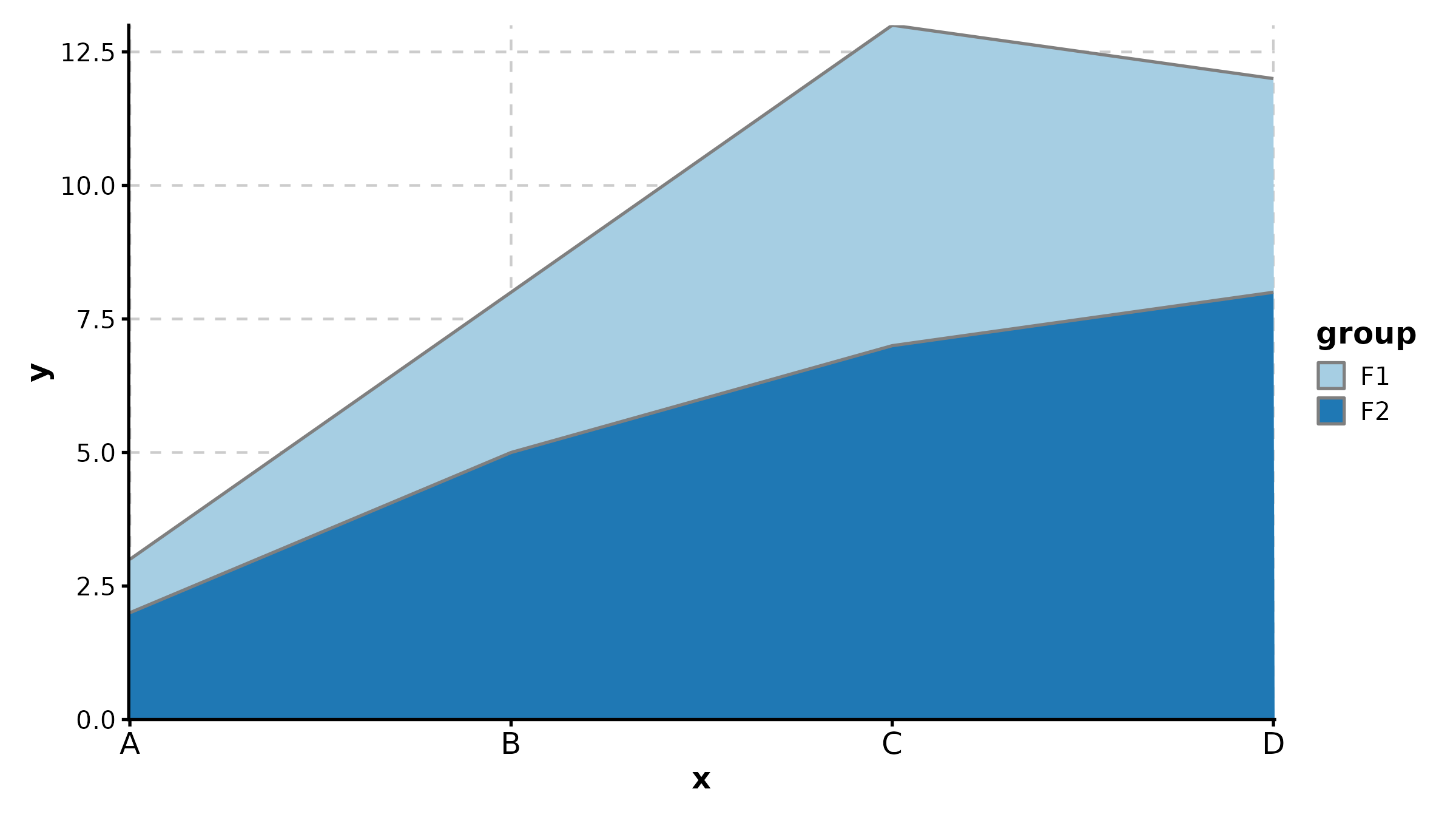
LollipopPlot
x is the numeric value axis, y is the
category axis.
lollipop_df <- data.frame(
pathway = paste0("Pathway ", LETTERS[1:8]),
score = c(3.2, 2.8, 2.5, 2.1, -1.5, -1.9, -2.3, -2.7)
)
LollipopPlot(lollipop_df, x = "score", y = "pathway",
palette = "Set2", title = "Lollipop Plot")
LollipopPlot(lollipop_df, x = "score", y = "pathway",
fill_by = "score", palette = "RdBu",
title = "Color by Score")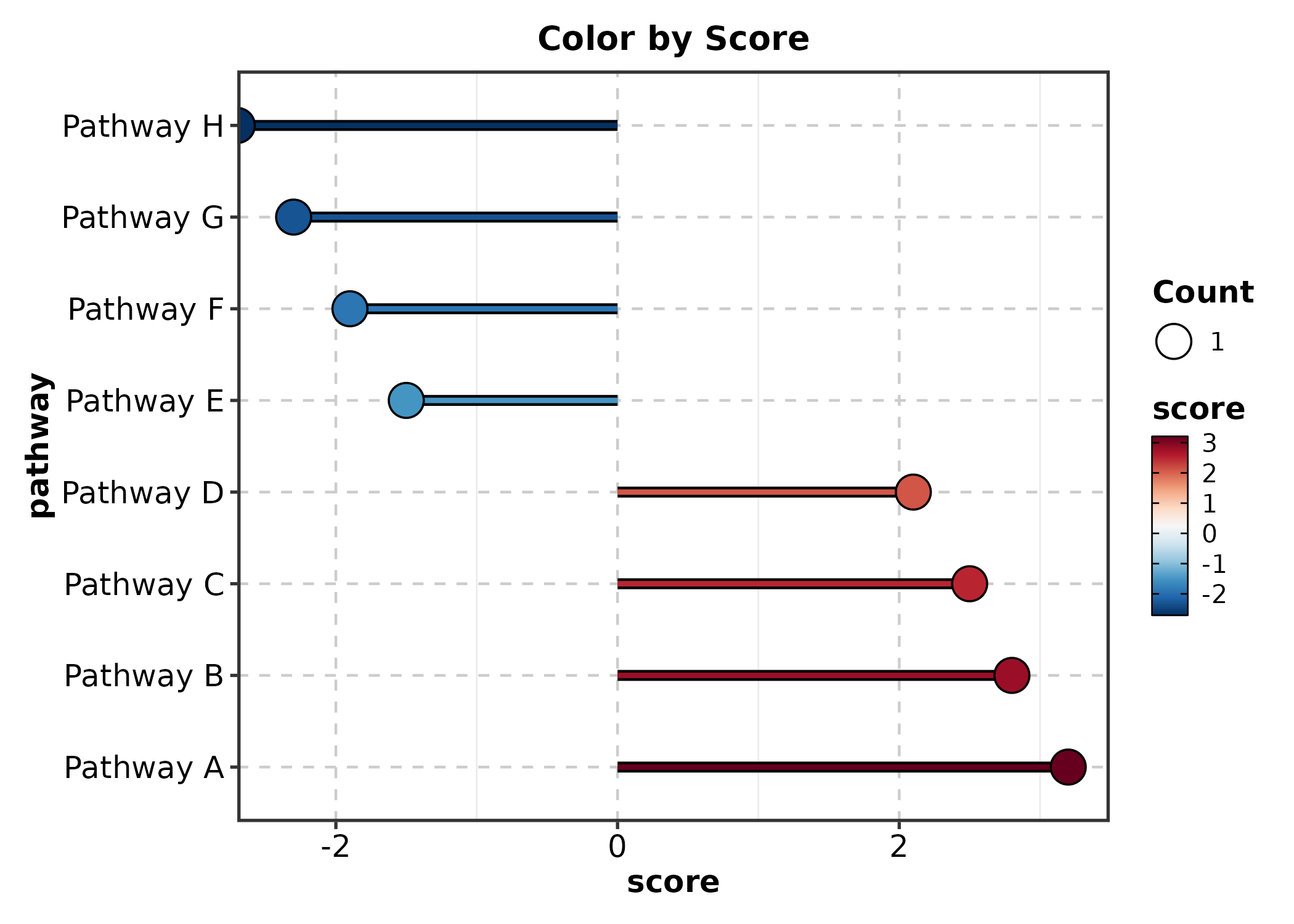
SplitBarPlot
x is the numeric value, y is the category
label. Positive and negative values are split automatically.
split_bar <- data.frame(
term = paste0("GO:", 1:10),
nes = c(2.1, 1.8, 1.5, 1.3, 1.1, -1.2, -1.4, -1.7, -1.9, -2.3)
)
SplitBarPlot(split_bar, x = "nes", y = "term",
palette = "RdBu", title = "NES Split Bar")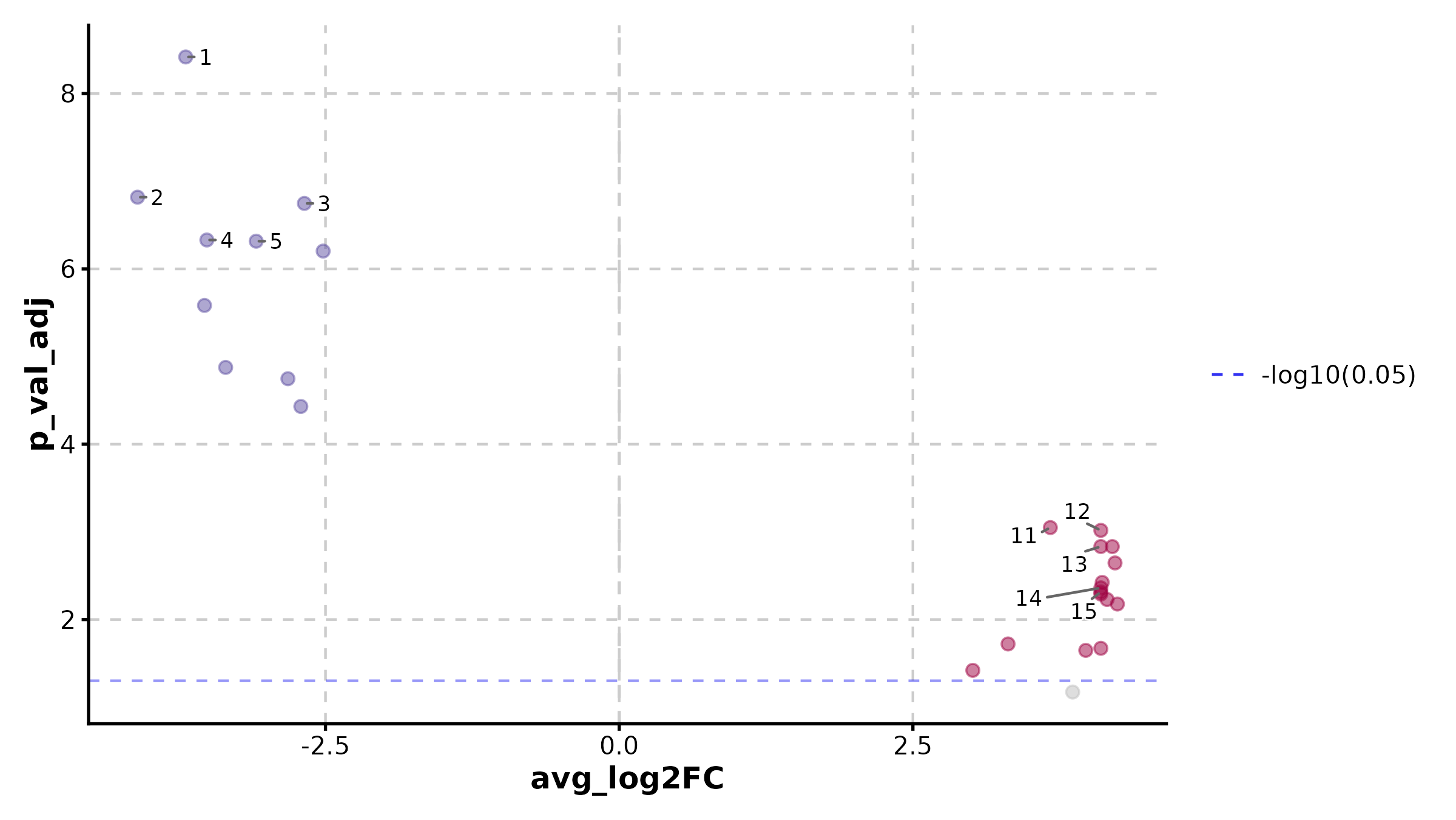
SplitBarPlot(split_bar, x = "nes", y = "term",
palette = "RdBu", flip = TRUE,
title = "Horizontal Split Bar")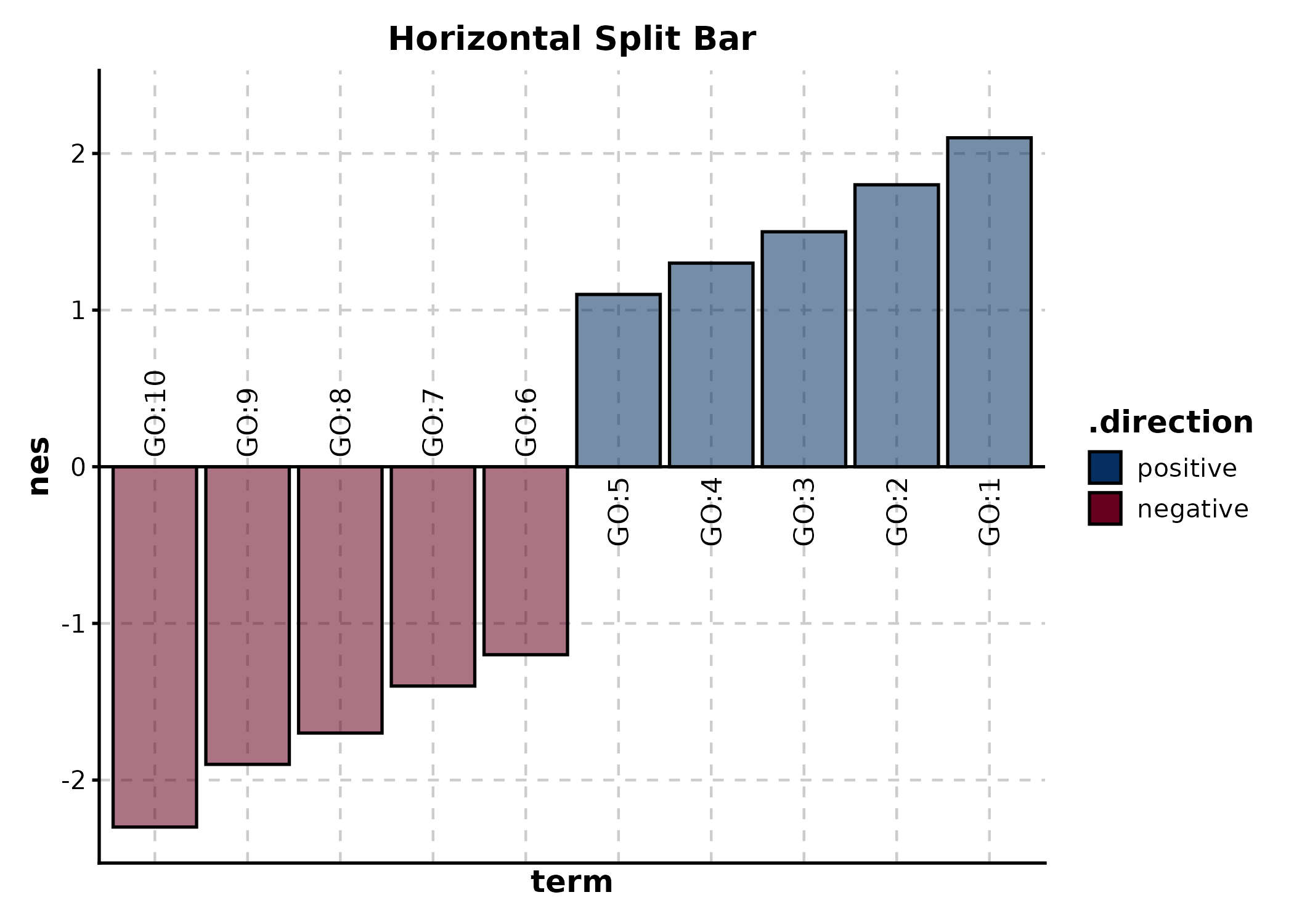
WaterfallPlot
Alias for SplitBarPlot. x = numeric,
y = category.
wf_df <- data.frame(
patient = paste0("Pt", 1:20),
change = sort(runif(20, -80, 40), decreasing = TRUE)
)
WaterfallPlot(wf_df, x = "change", y = "patient",
palette = "RdBu", title = "Tumor Size Change (%)")
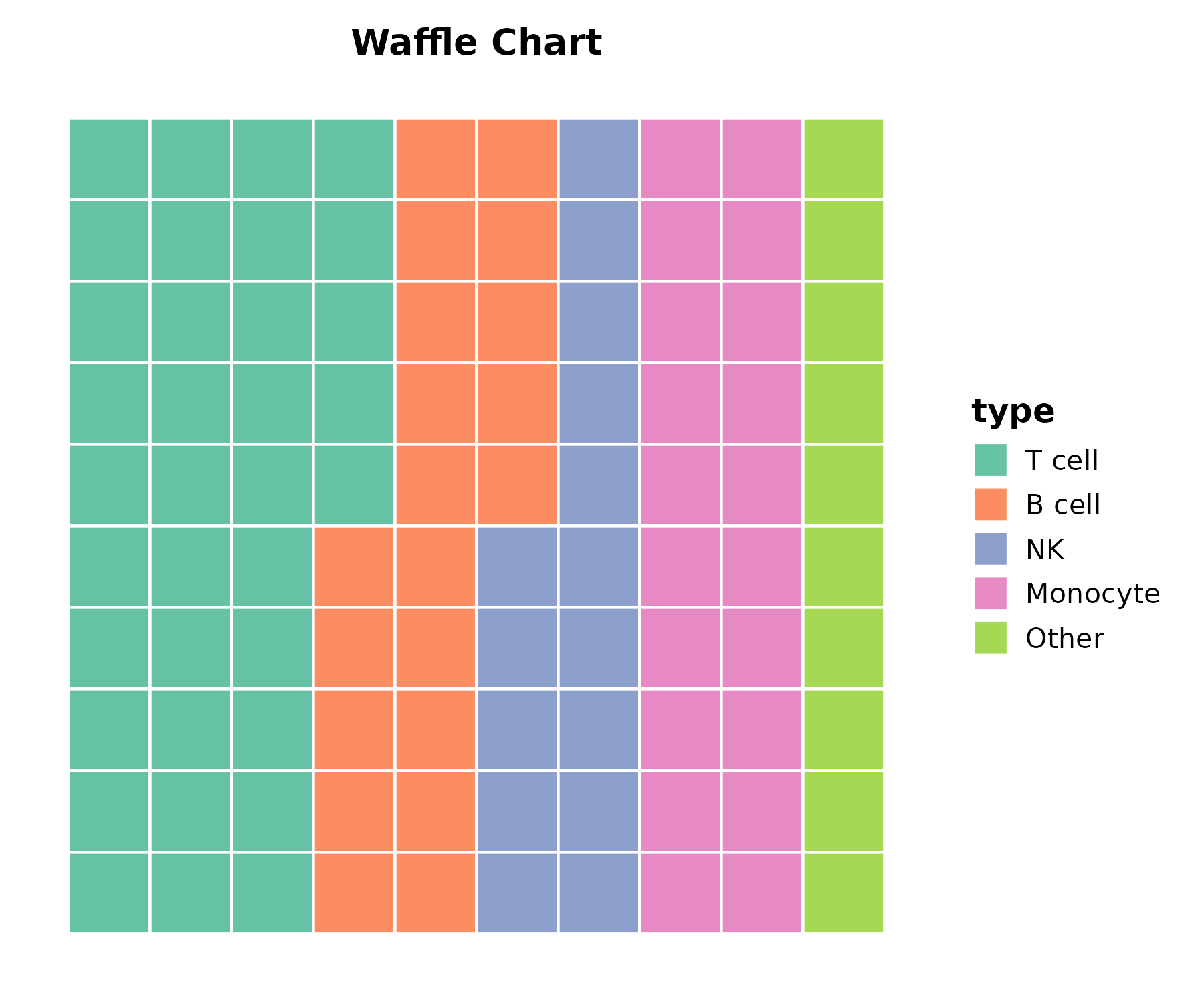
PieChart
cell_comp <- data.frame(
type = c("T cell", "B cell", "NK", "Monocyte", "Other"),
count = c(35, 20, 15, 20, 10)
)
PieChart(cell_comp, x = "type", y = "count", palette = "Set2",
title = "Cell Composition")
RingPlot
RingPlot(cell_comp, x = "type", y = "count", group_by = "type",
palette = "Set2", title = "Ring Chart")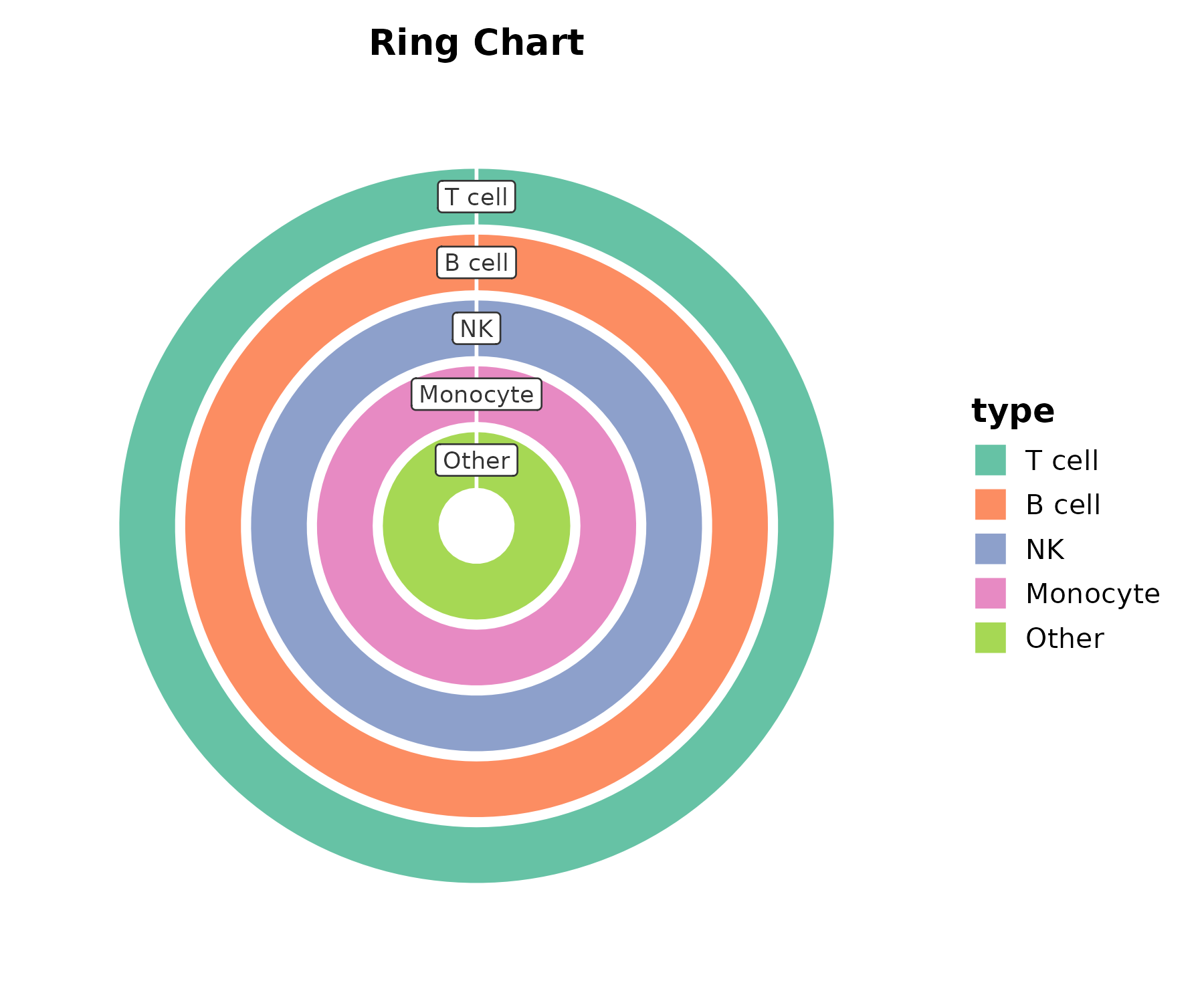
RadarPlot
radar_df <- data.frame(
metric = rep(c("Proliferation", "Apoptosis", "Migration",
"Invasion", "Angiogenesis"), 2),
value = c(8, 4, 7, 3, 6, 5, 8, 4, 9, 3),
group = rep(c("Wild Type", "Mutant"), each = 5)
)
RadarPlot(radar_df, x = "metric", y = "value", group_by = "group",
scale_y = "none", palette = "Set1", title = "Radar Plot")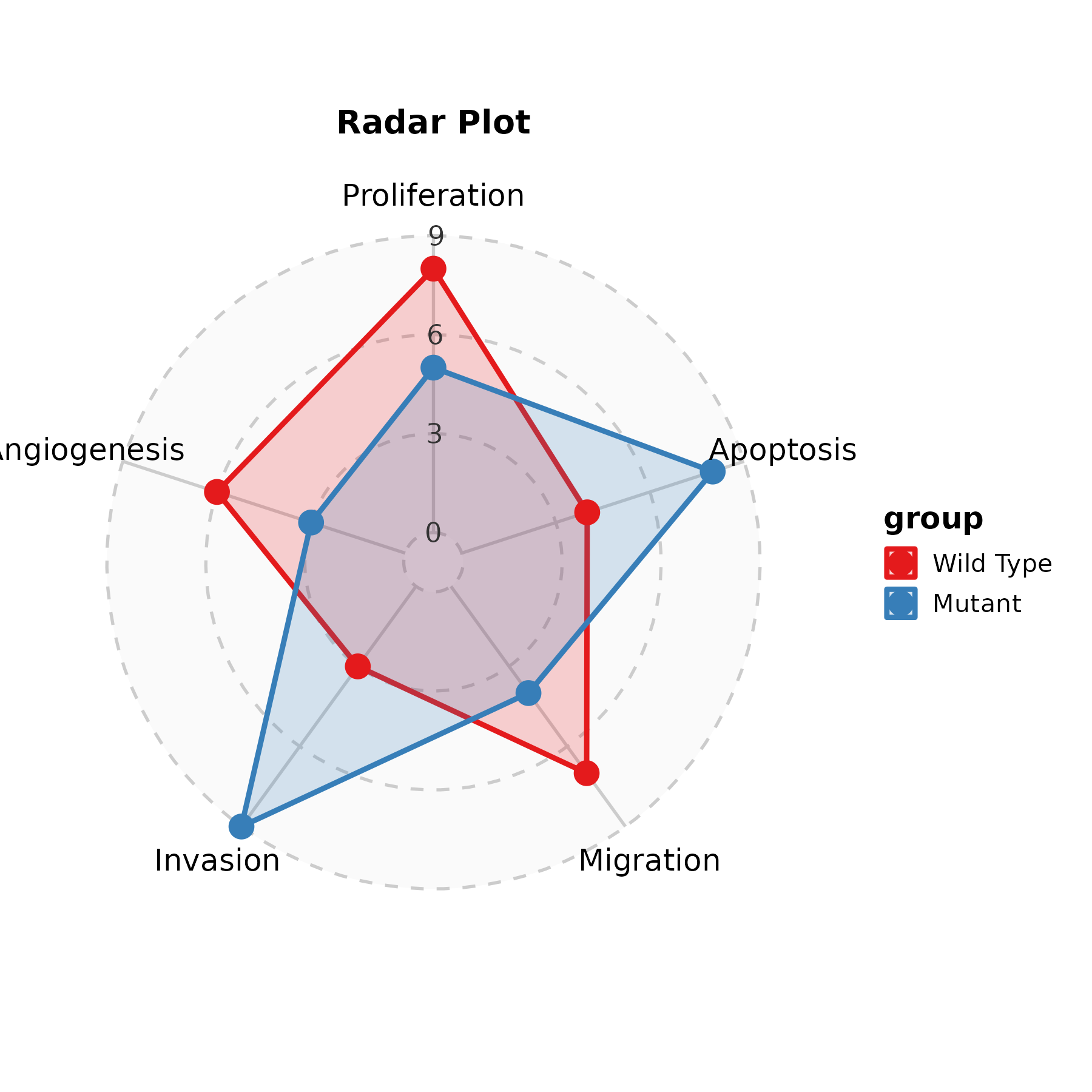
SpiderPlot
SpiderPlot(radar_df, x = "metric", y = "value", group_by = "group",
scale_y = "none", palette = "lancet", title = "Spider Plot")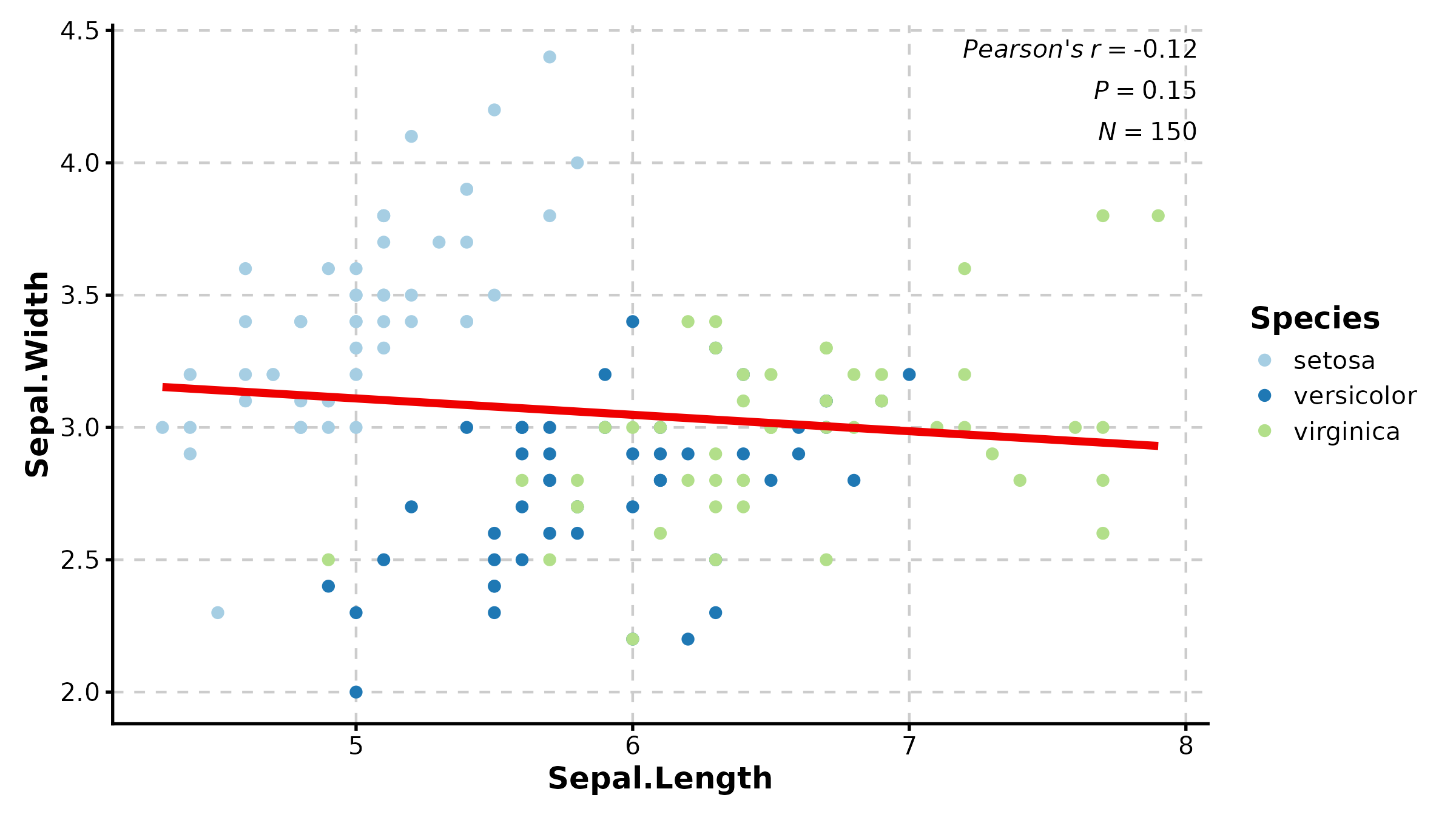
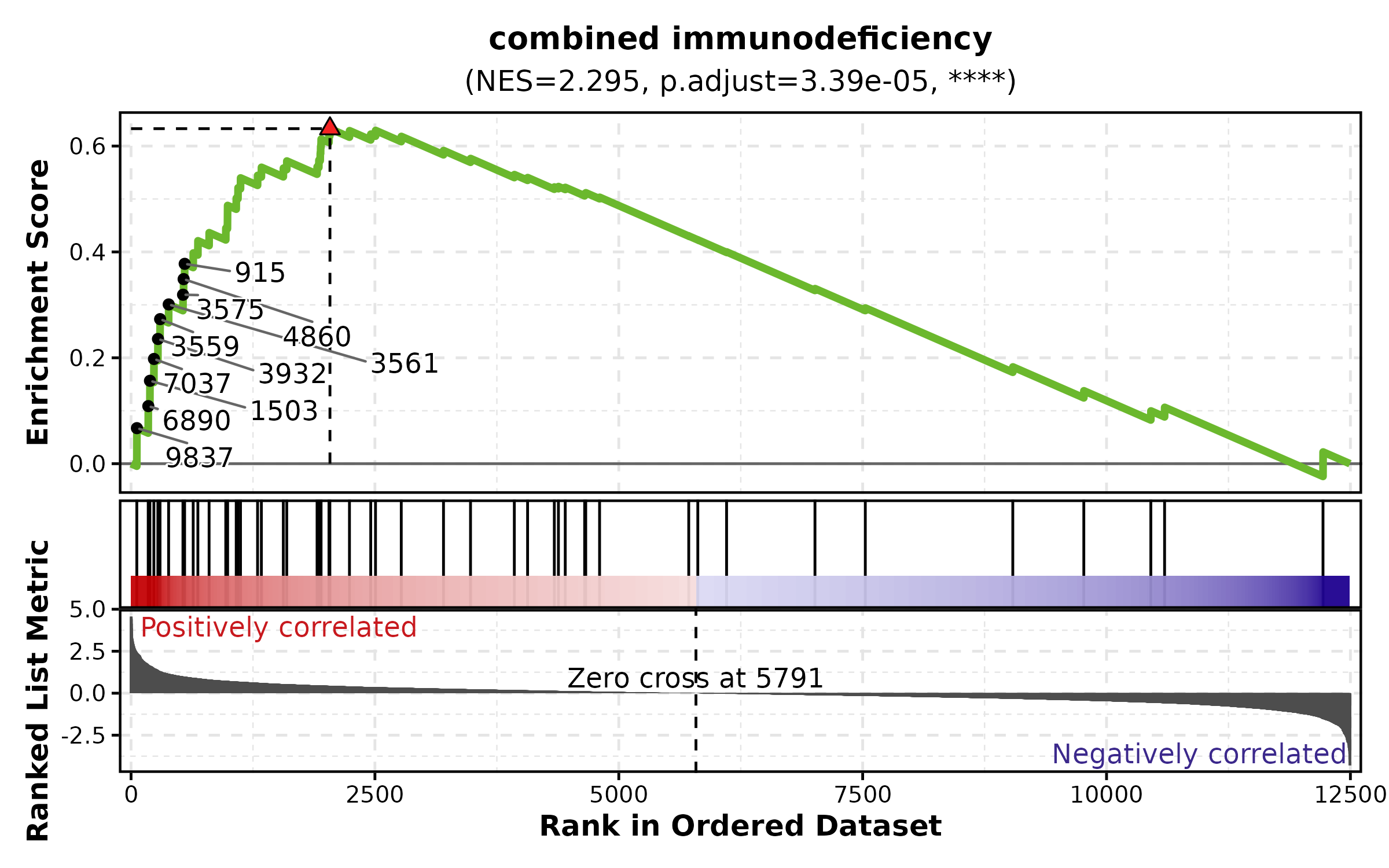
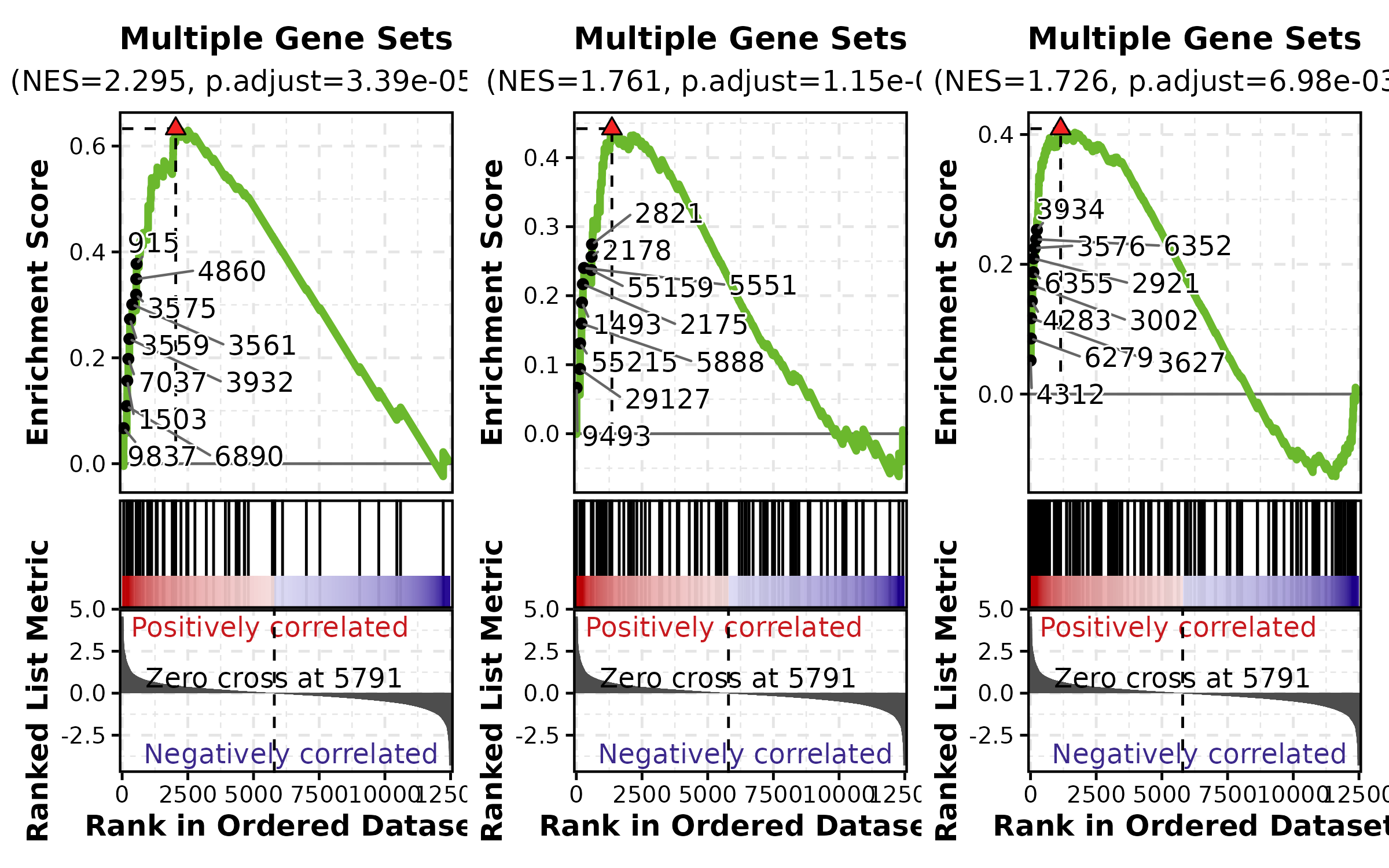
2. Enrichment & Pathway
EnrichMap
data(enrich_example)
EnrichMap(enrich_example, top_term = 20, layout = "fr",
palette = "Spectral", title = "GO Enrichment Map")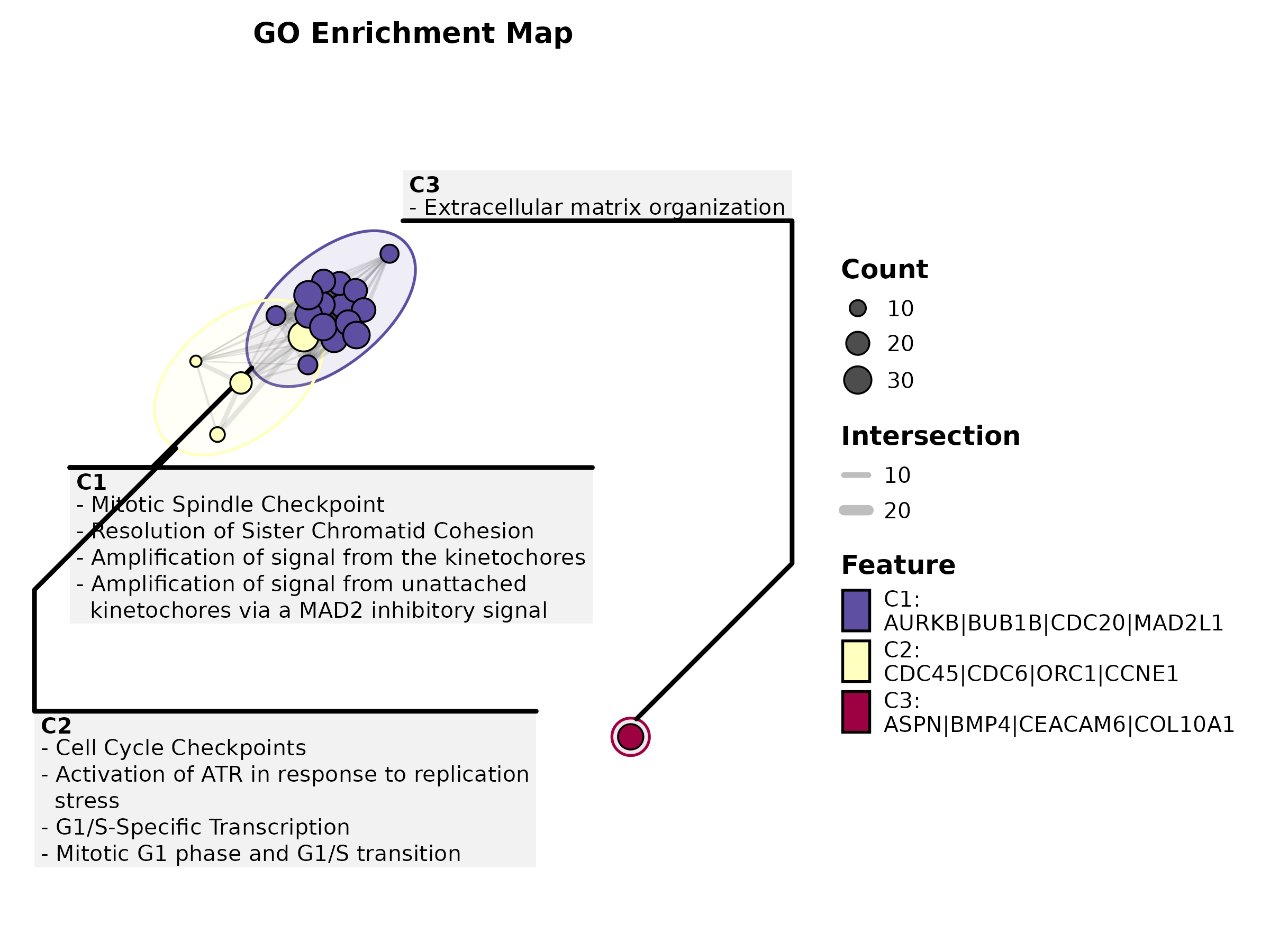
EnrichMap(enrich_example, top_term = 15, show_keyword = TRUE,
label = "term", title = "With Keyword Clustering")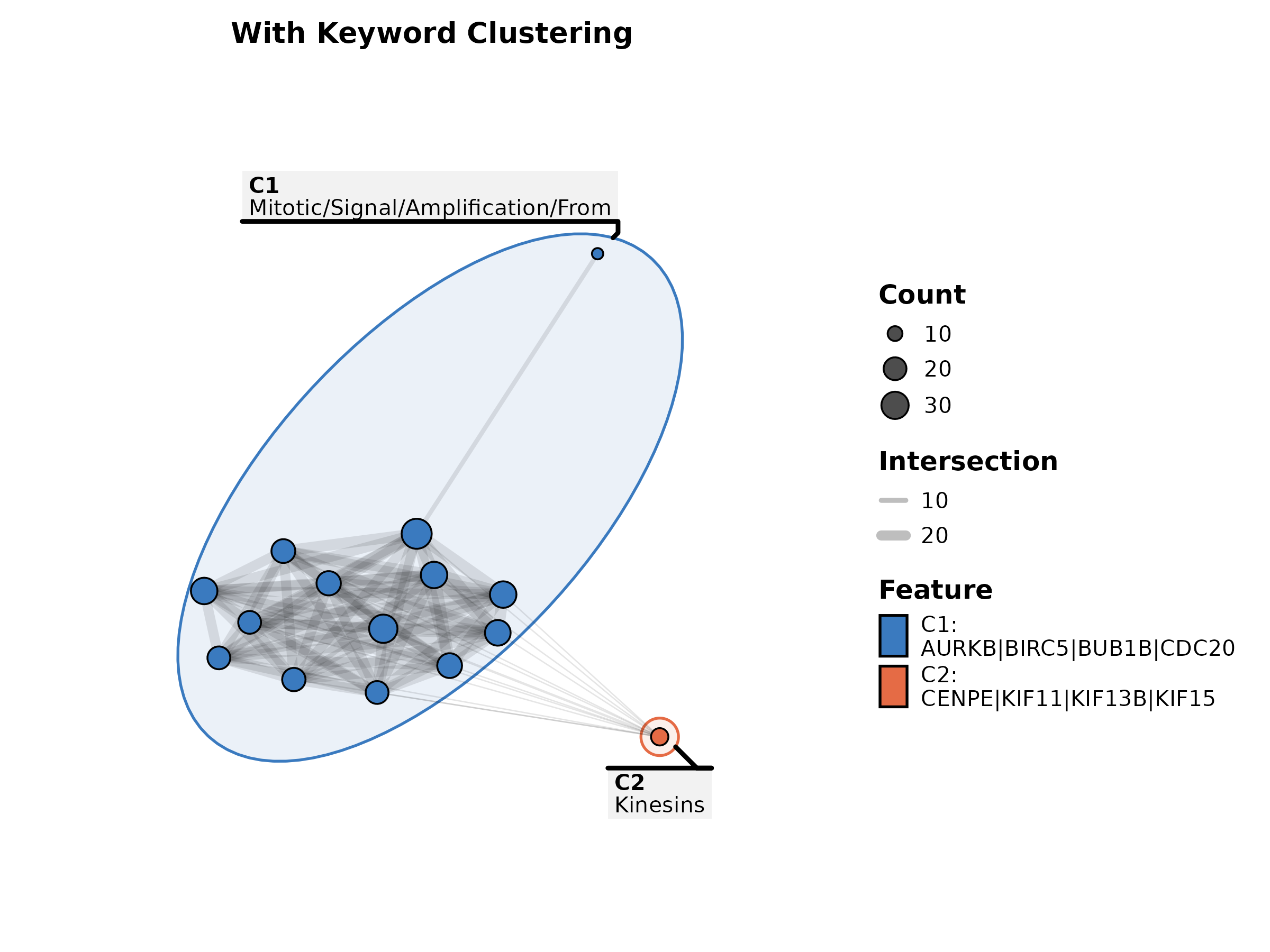
EnrichNetwork
data(enrich_multidb_example)
EnrichNetwork(enrich_multidb_example, top_term = 15, layout = "fr",
palette = "Set3", title = "Enrichment Network")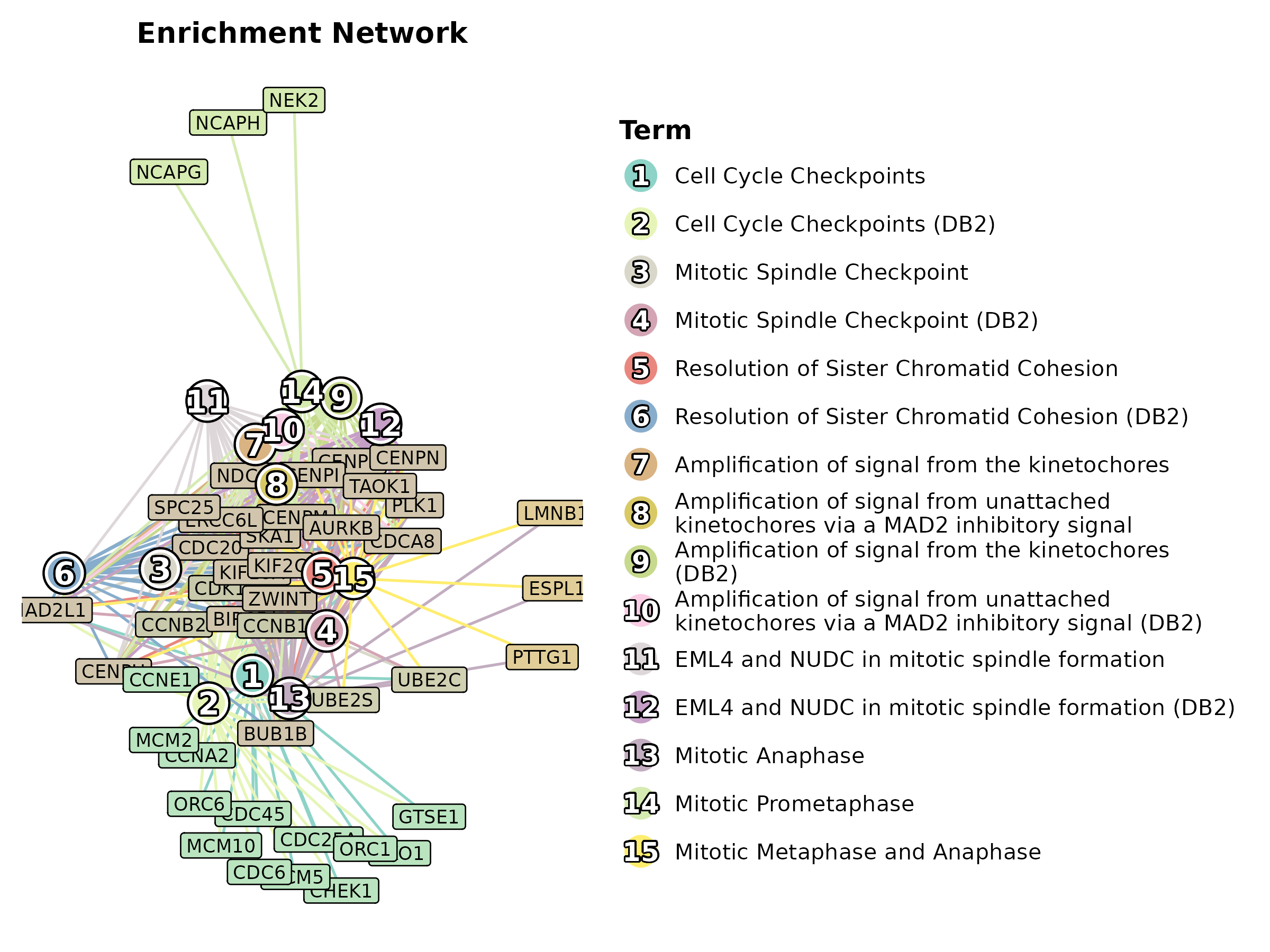
EnrichNetwork(enrich_multidb_example, top_term = 20, layout = "circle",
palette = "Paired", title = "Circular Layout")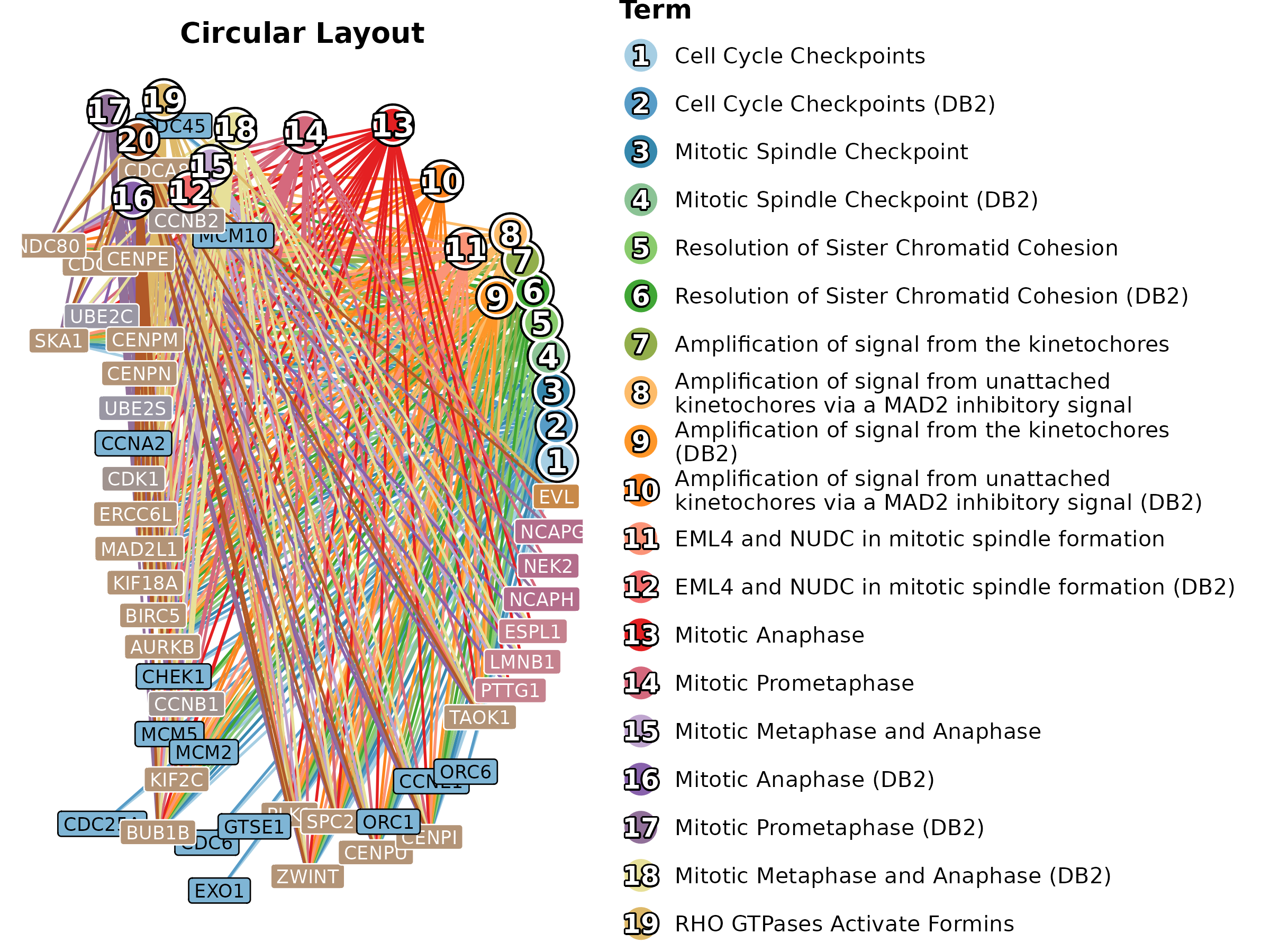
GSEASummaryPlot
data(gsea_example)
GSEASummaryPlot(gsea_example, top_term = 15, palette = "RdBu",
title = "GSEA Summary")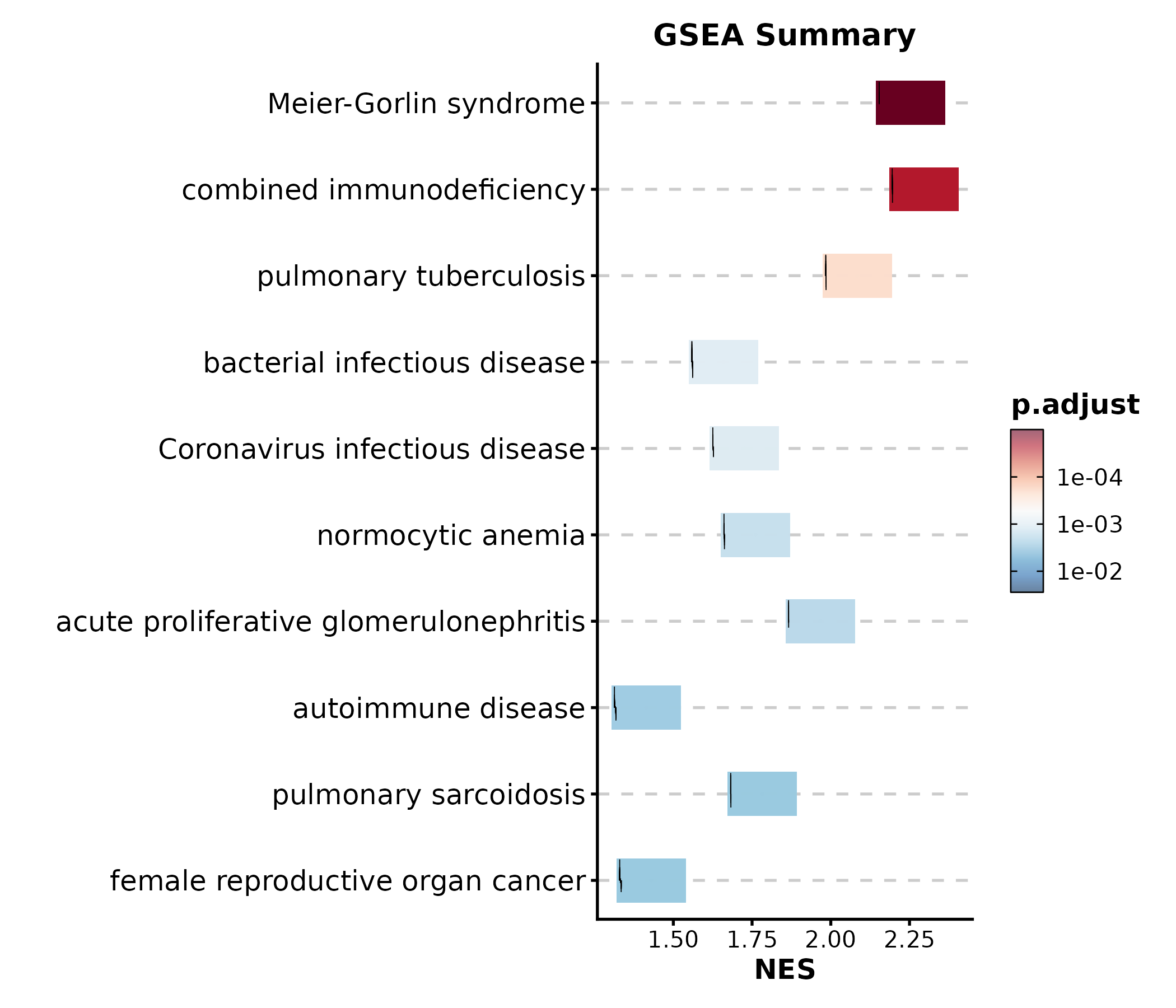
GSEASummaryPlot(gsea_example, top_term = 10, palette = "PiYG",
title = "Top 10 Pathways")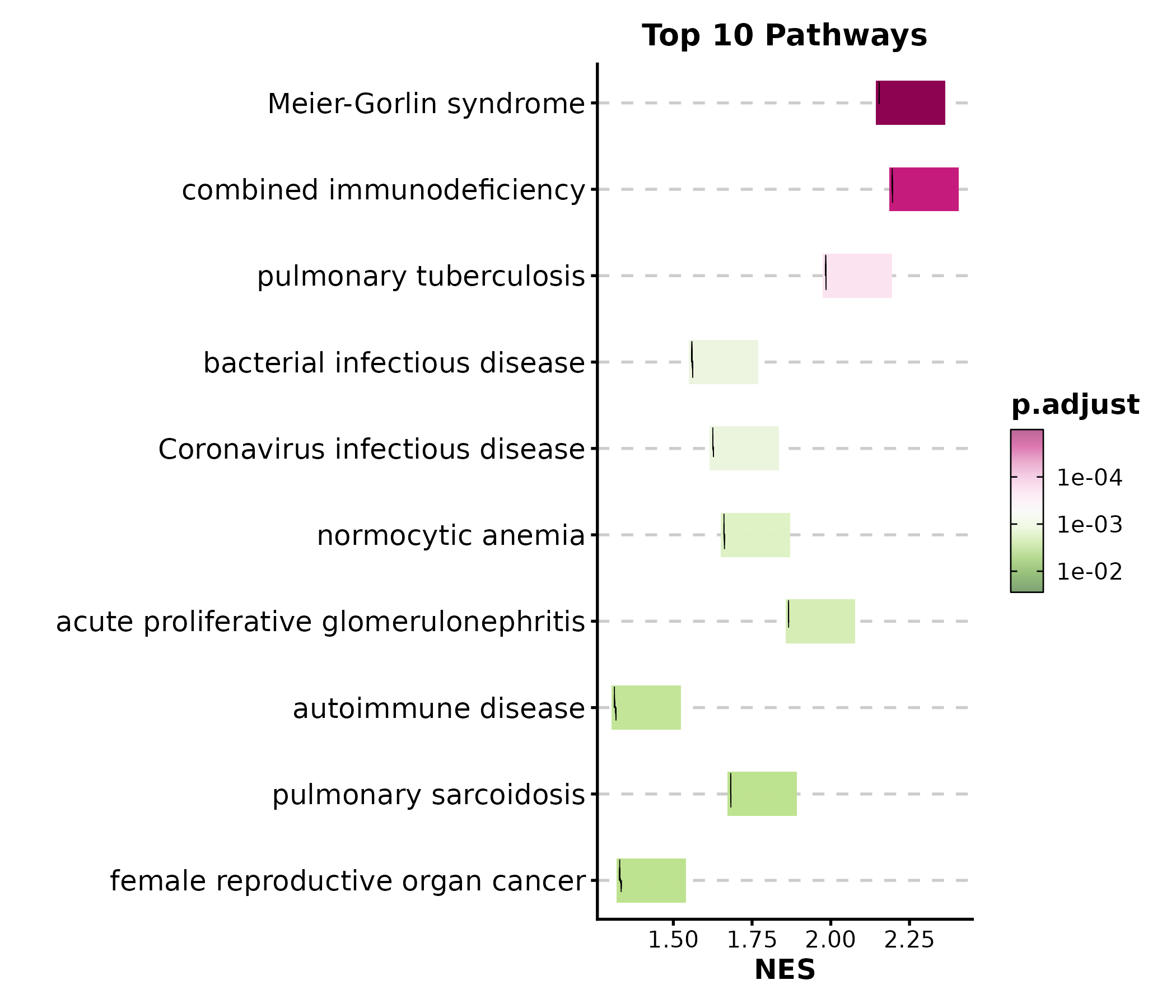
3. Single-Cell & Spatial
DimPlot
data(dim_example)
DimPlot(dim_example, dims = c("basis_1", "basis_2"),
group_by = "clusters", palette = "igv",
pt_size = 1.2, label = TRUE, label_insitu = TRUE,
title = "UMAP Clustering")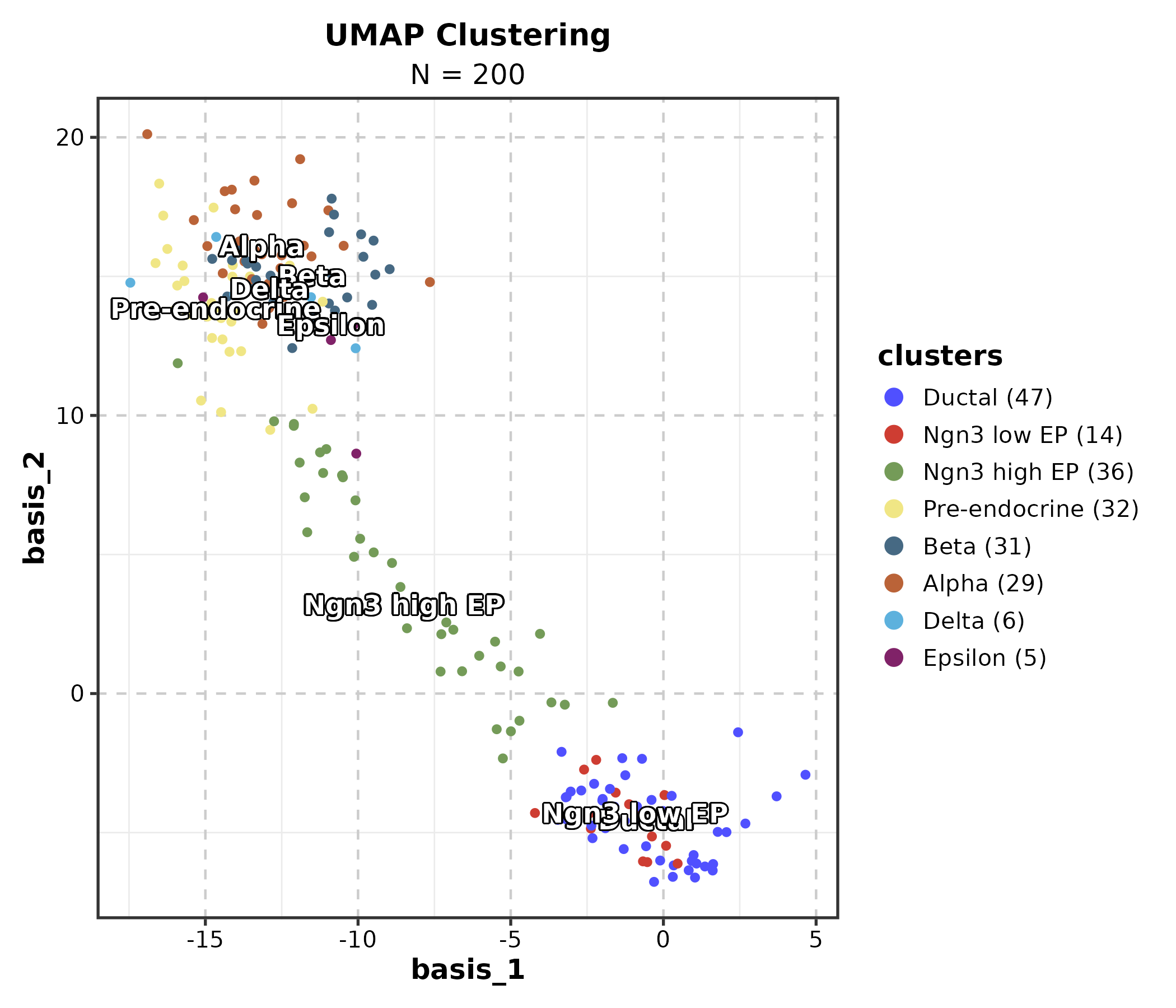
DimPlot(dim_example, dims = c("basis_1", "basis_2"),
group_by = "clusters", palette = "Set3",
add_density = TRUE, title = "With Density Contours")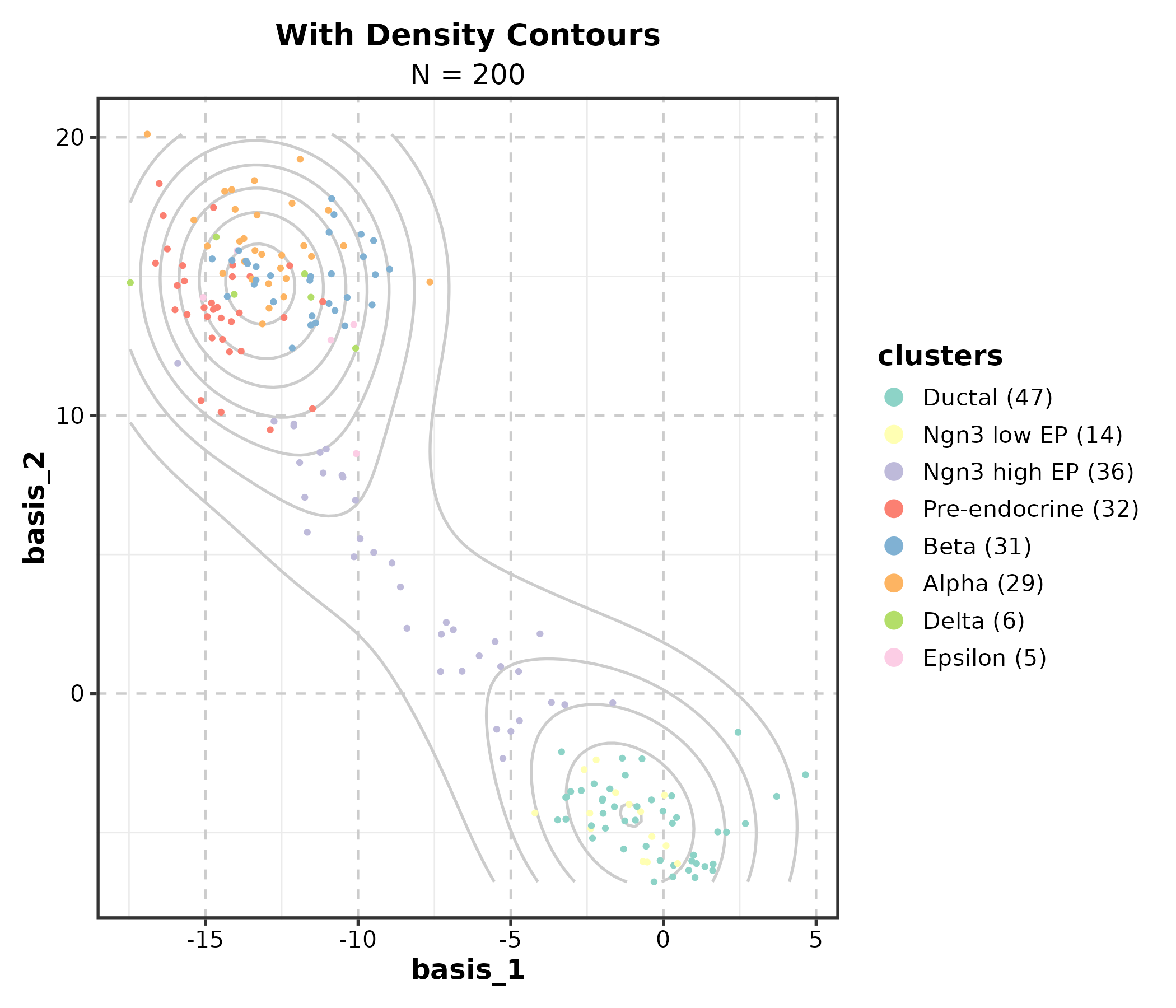
DimPlot(dim_example, dims = c("basis_1", "basis_2"),
group_by = "clusters", palette = "igv",
split_by = "group", combine = TRUE, nrow = 1,
title = "Split by Group")
FeatureDimPlot
dim_example$CD3D <- rnorm(nrow(dim_example))
FeatureDimPlot(dim_example, dims = c("basis_1", "basis_2"),
features = "CD3D", palette = "viridis", pt_size = 1.2,
title = "Feature Expression")
FeatureDimPlot(dim_example, dims = c("basis_1", "basis_2"),
features = "CD3D", palette = "RdBu", pt_size = 1.2,
title = "Diverging Palette")
VelocityPlot
embedding <- as.matrix(dim_example[, c("basis_1", "basis_2")])
v_embedding <- as.matrix(dim_example[, c("stochasticbasis_1", "stochasticbasis_2")])
VelocityPlot(embedding = embedding, v_embedding = v_embedding,
plot_type = "grid", title = "RNA Velocity Field")
TrajectoryPlot
dim_example$pseudotime <- abs(rnorm(nrow(dim_example)))
TrajectoryPlot(dim_example, dims = c("basis_1", "basis_2"),
pseudotime = "pseudotime", group_by = "clusters",
palette = "viridis", title = "Pseudotime Trajectory")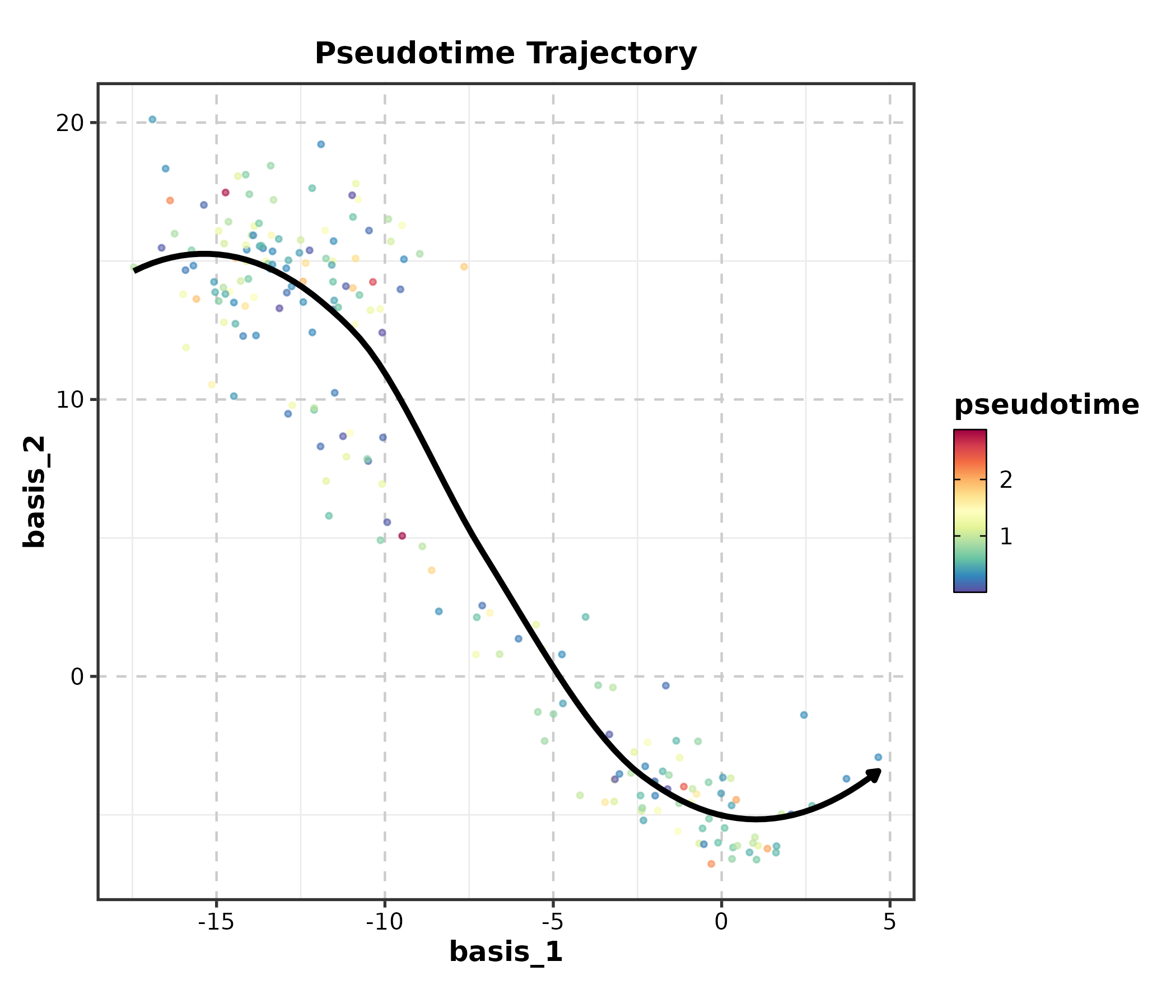
StackedViolinPlot
marker_df <- data.frame(
cluster = sample(c("T cell", "B cell", "NK", "Mono"), 200, replace = TRUE),
CD3D = rnorm(200, 3), CD8A = rnorm(200, 2),
MS4A1 = rnorm(200, 4), NKG7 = rnorm(200, 1)
)
StackedViolinPlot(marker_df,
features = c("CD3D", "CD8A", "MS4A1", "NKG7"),
group_by = "cluster", palette = "npg",
title = "Marker Gene Expression")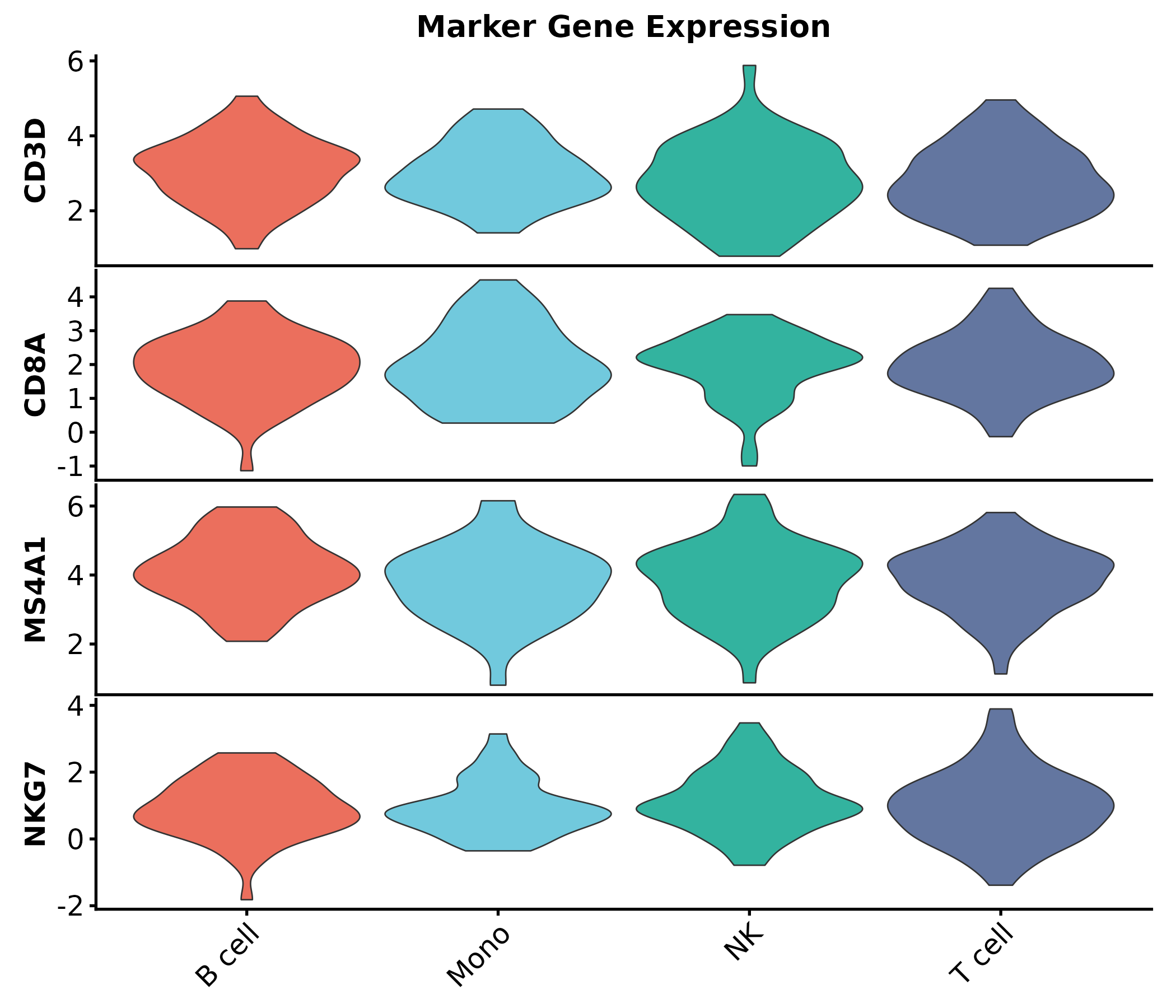
ClustreePlot
Requires clustree and a Seurat object or data frame with
resolution columns.
ClustreePlot(seurat_obj, prefix = "RNA_snn_res.",
title = "Clustering Resolution Tree")SpatImagePlot / SpatPointsPlot / SpatShapesPlot / SpatMasksPlot
These require spatial data objects (SpatRaster, SpatVector). See
?SpatImagePlot for details.
SpatImagePlot(spe, feature = "gene_of_interest",
palette = "viridis", title = "Spatial Expression")
SpatPointsPlot(spe, group_by = "celltype",
palette = "igv", title = "Spatial Cell Types")
SpatShapesPlot(spe, group_by = "region",
palette = "Set3", title = "Spatial Regions")
SpatMasksPlot(spe, group_by = "mask_type",
palette = "Set2", title = "Spatial Masks")4. Genomics
VolcanoPlot
real_genes <- c("TP53","EGFR","MYC","KRAS","BRCA1","CDK4","PTEN","RB1",
"PIK3CA","AKT1","CD274","CTLA4","PDCD1","IDO1","VEGFA",
"BRAF","JAK2","STAT3","NRAS","HAVCR2")
deg <- data.frame(
gene = c(real_genes, paste0("Gene", 1:480)),
log2FC = c(rnorm(10, 2.5, 0.8), rnorm(10, -2.5, 0.8), rnorm(480, 0, 0.8)),
padj = c(runif(20, 1e-10, 1e-3), runif(480, 0.01, 1))
)
VolcanoPlot(deg, x = "log2FC", y = "padj", label_by = "gene",
x_cutoff = 1, y_cutoff = 0.05, nlabel = 10,
title = "Volcano Plot")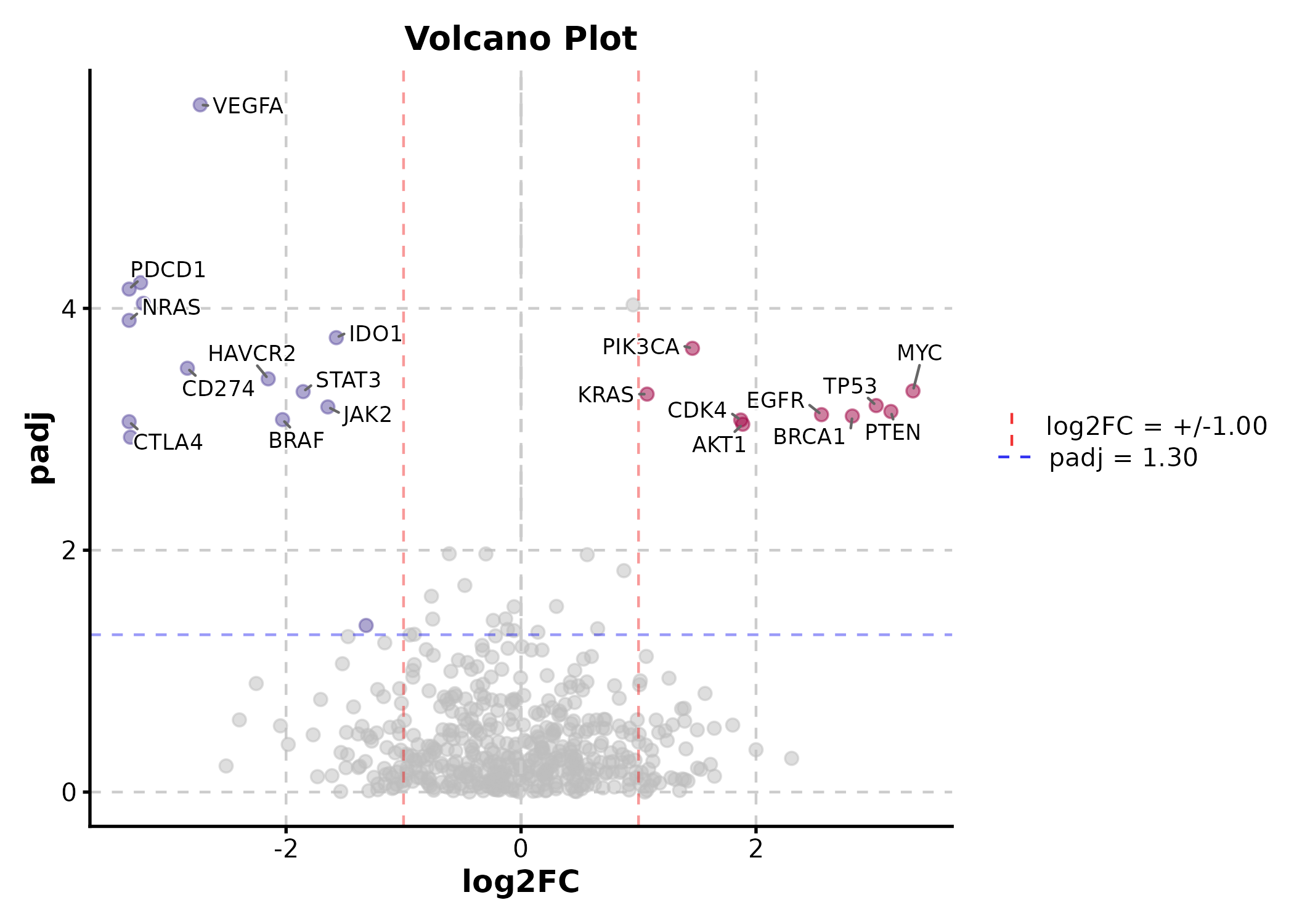
VolcanoPlot(deg, x = "log2FC", y = "padj", label_by = "gene",
x_cutoff = 1, y_cutoff = 0.05, nlabel = 5,
flip_negatives = TRUE, title = "Mirrored Volcano")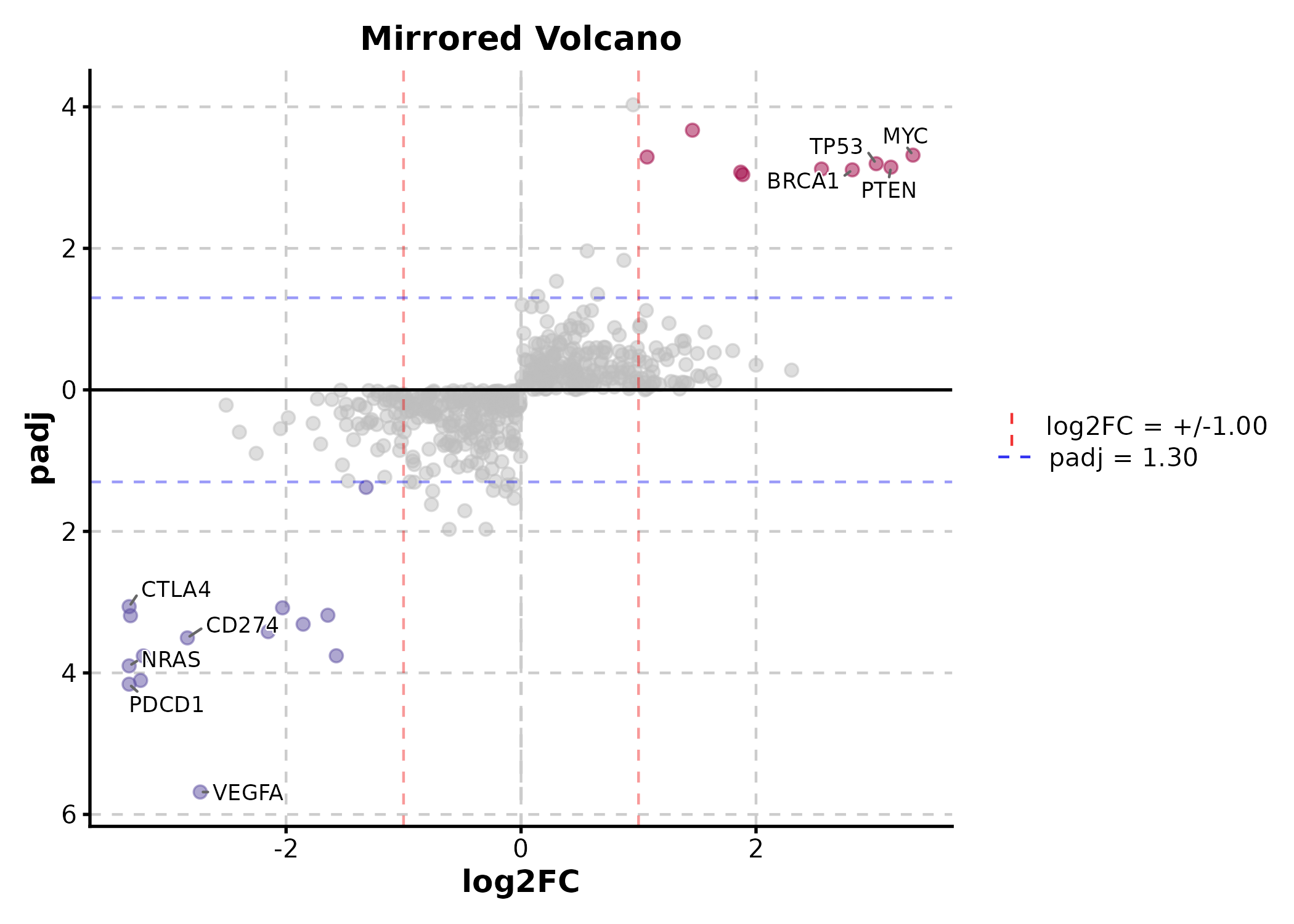
deg$group <- sample(c("Responder", "NonResponder"), 500, replace = TRUE)
VolcanoPlot(deg, x = "log2FC", y = "padj", label_by = "gene",
x_cutoff = 1, y_cutoff = 0.05, nlabel = 5,
facet_by = "group", title = "Faceted Volcano")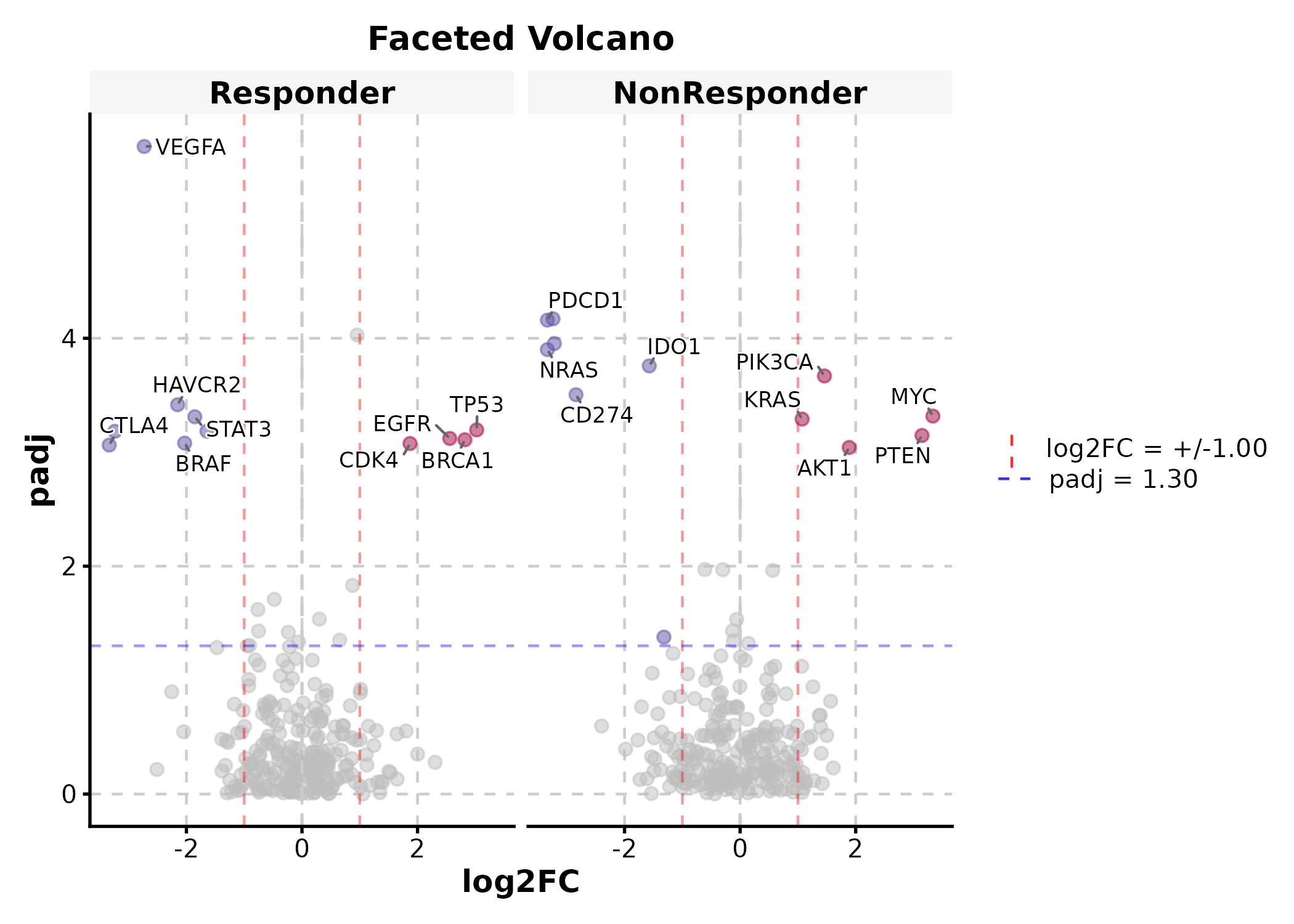
ManhattanPlot
gwas <- data.frame(
chr = rep(paste0("chr", 1:22), each = 500),
pos = rep(1:500, 22) * 1e5,
pvalue = runif(11000, 0, 1)
)
gwas$pvalue[sample(11000, 30)] <- runif(30, 0, 1e-8)
ManhattanPlot(gwas, chr_by = "chr", pos_by = "pos", pval_by = "pvalue",
signif = 5e-8, title = "GWAS Results")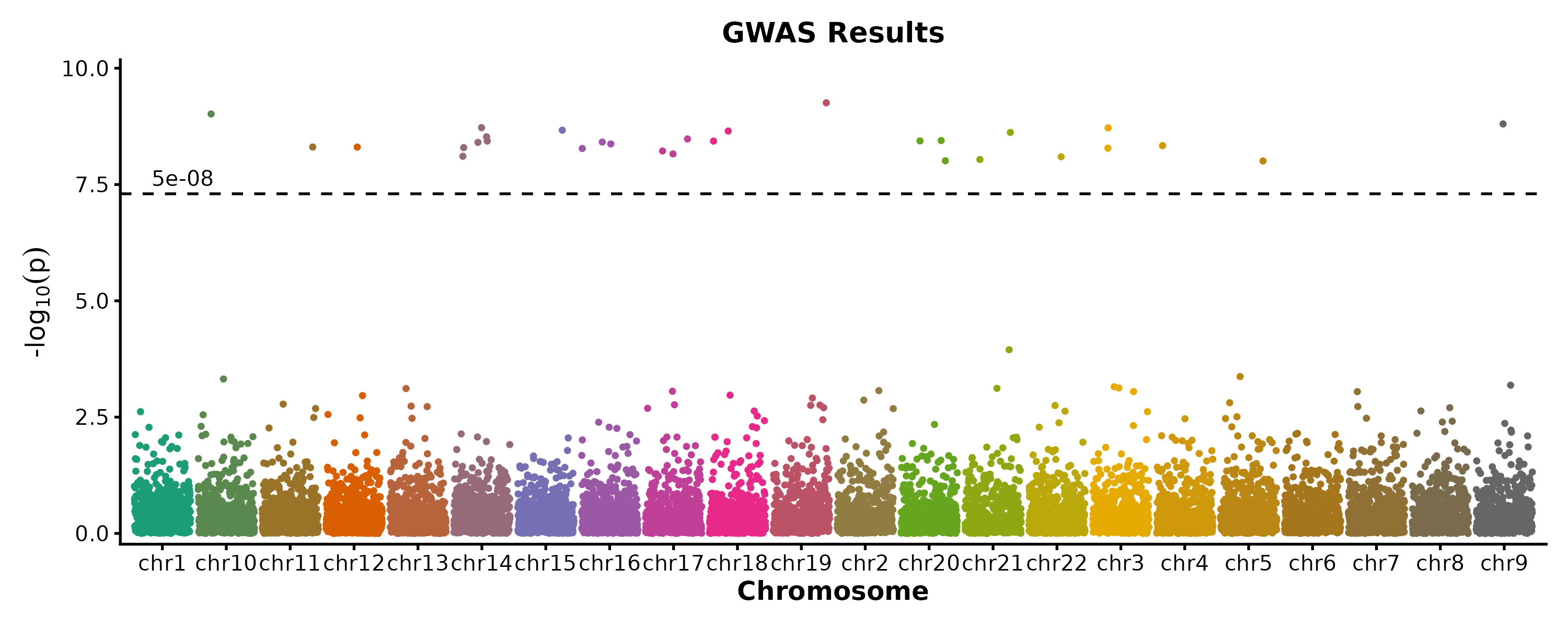
VennDiagram
gene_sets <- list(
"RNA-seq" = paste0("Gene", 1:100),
"Proteomics" = paste0("Gene", 50:150),
"Metabolomics" = paste0("Gene", 80:160)
)
VennDiagram(gene_sets, palette = "Set2", title = "Multi-Omics Overlap")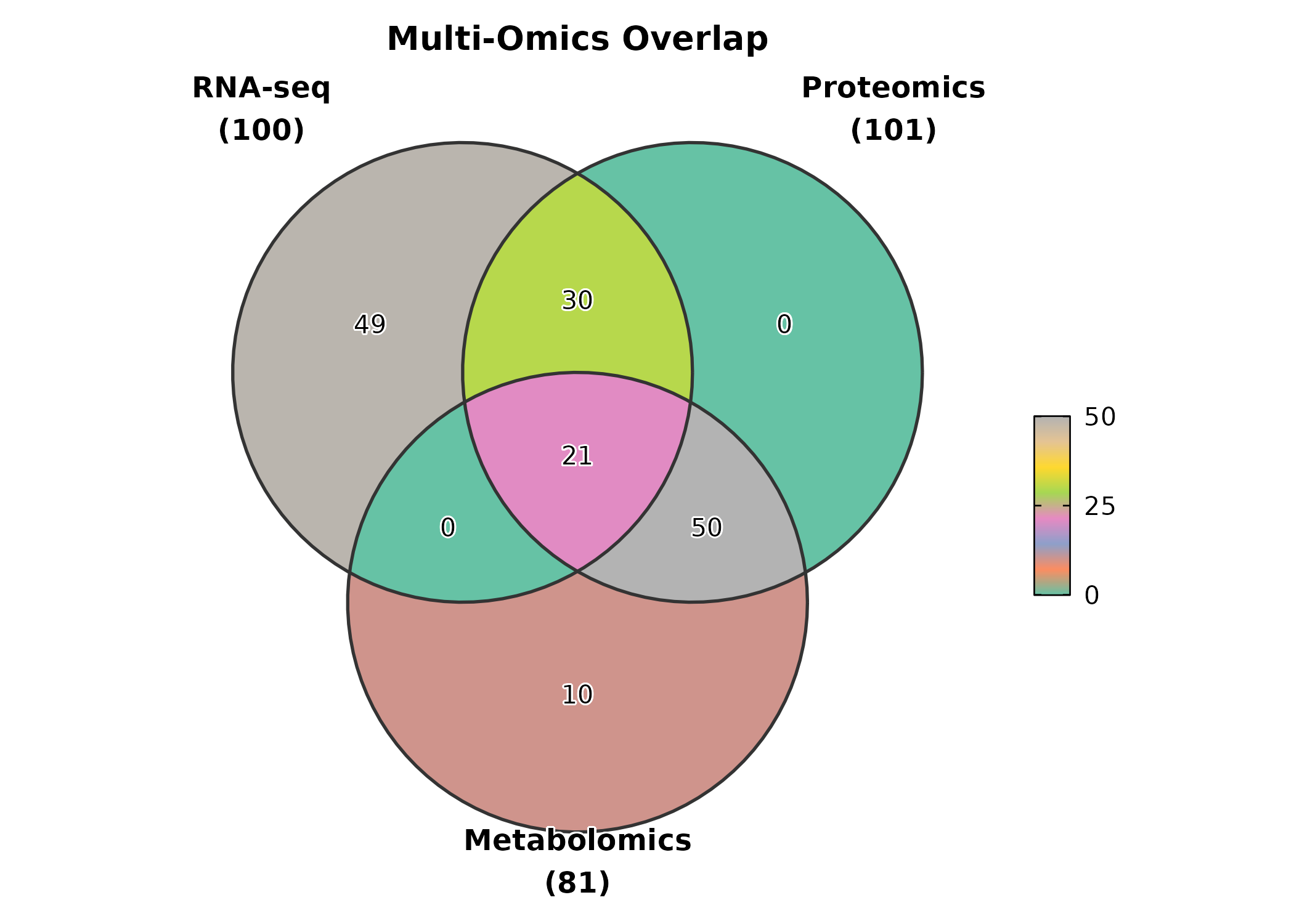
VennDiagram(gene_sets, palette = "material-indigo",
label = "both", alpha = 0.6,
title = "With Count + Percentage")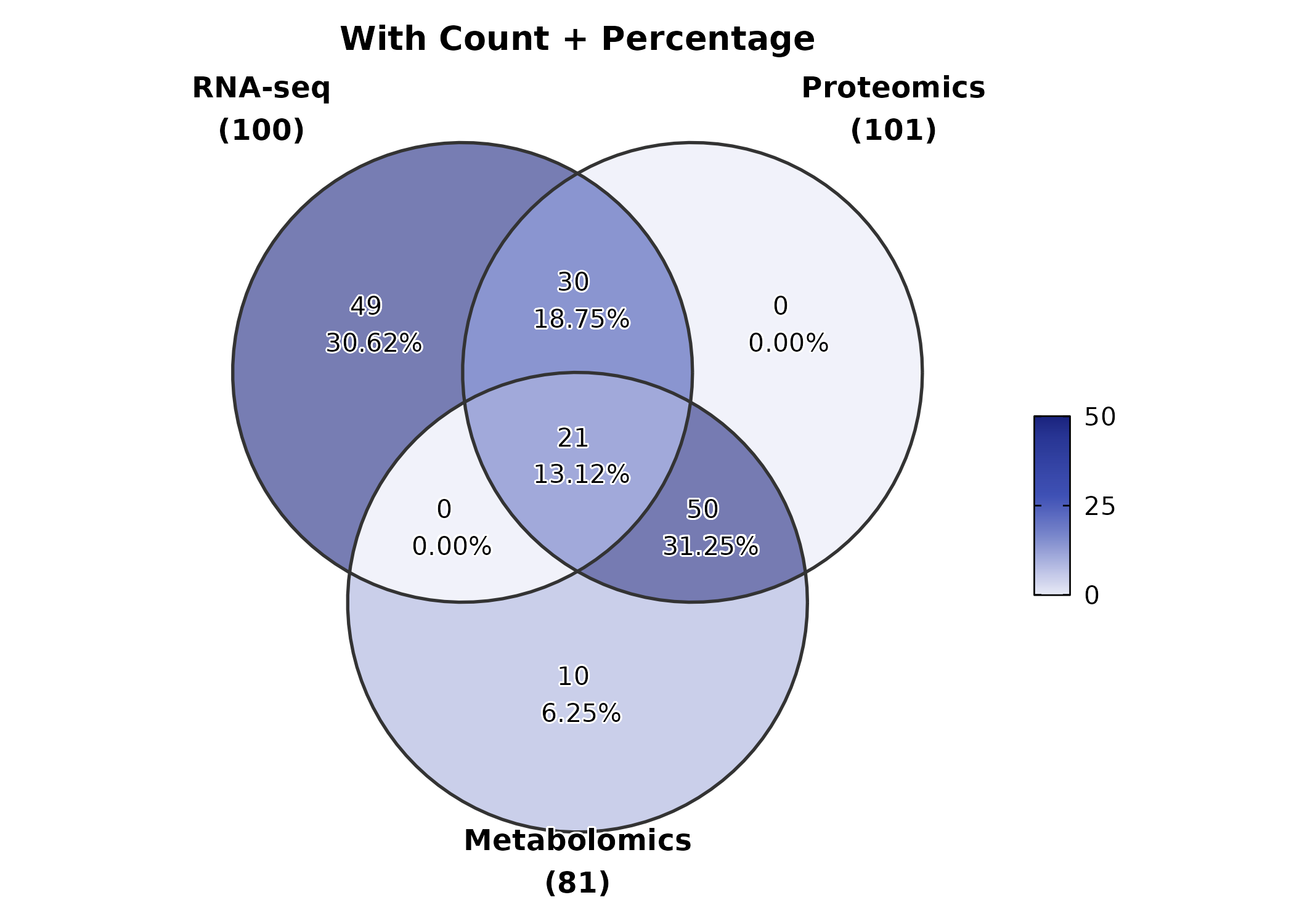
5. Clinical & Prediction
KMPlot
surv <- data.frame(
time = c(rexp(80, 0.008), rexp(70, 0.015)),
status = sample(0:1, 150, replace = TRUE, prob = c(0.35, 0.65)),
risk = rep(c("Low", "High"), c(80, 70))
)
KMPlot(surv, time = "time", status = "status", group_by = "risk",
palette = "jco", show_risk_table = TRUE, show_pval = TRUE,
title = "Kaplan-Meier Curve")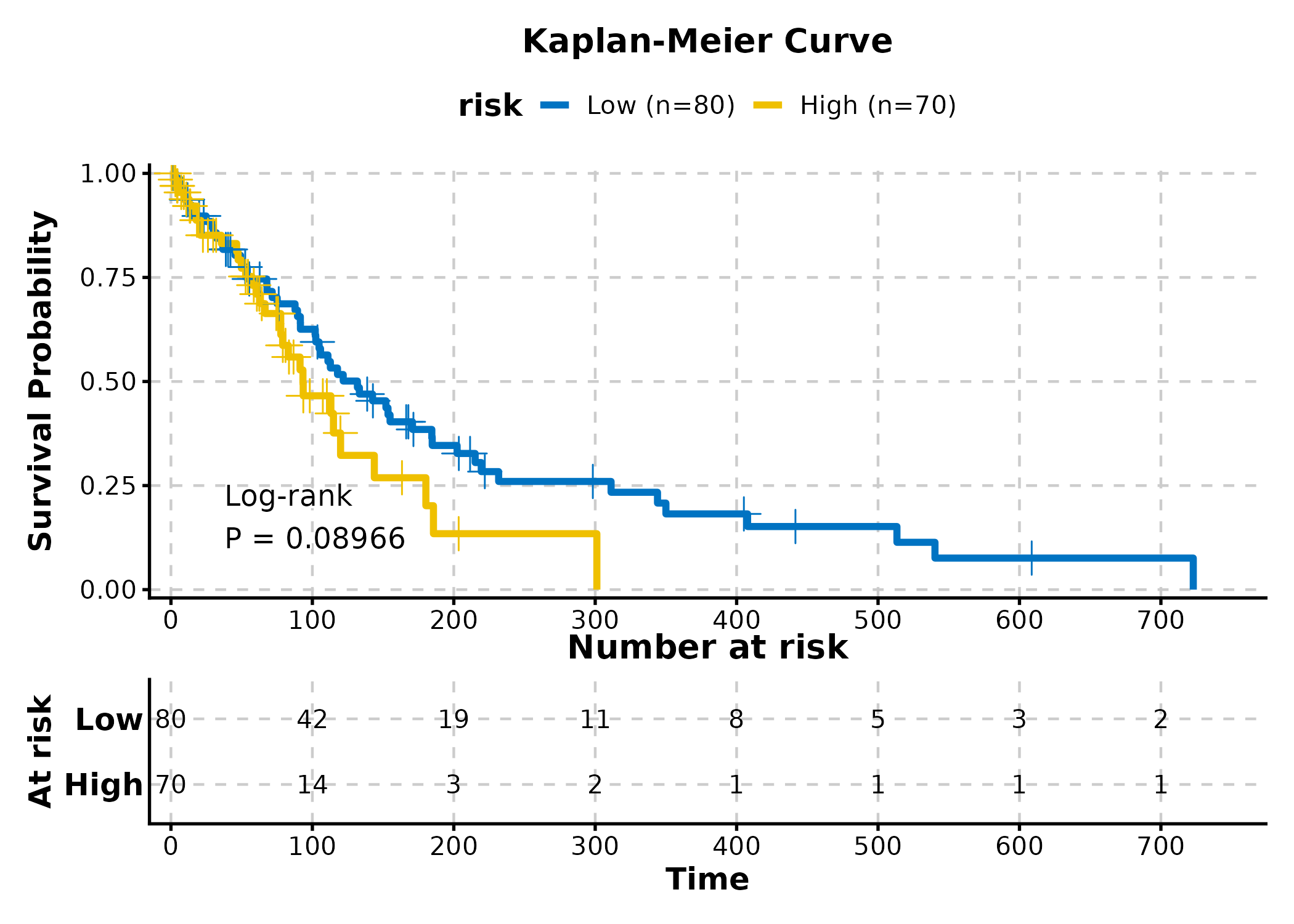
KMPlot(surv, time = "time", status = "status", group_by = "risk",
palette = "lancet", show_conf_int = TRUE,
show_median_line = "hv", title = "With CI and Median")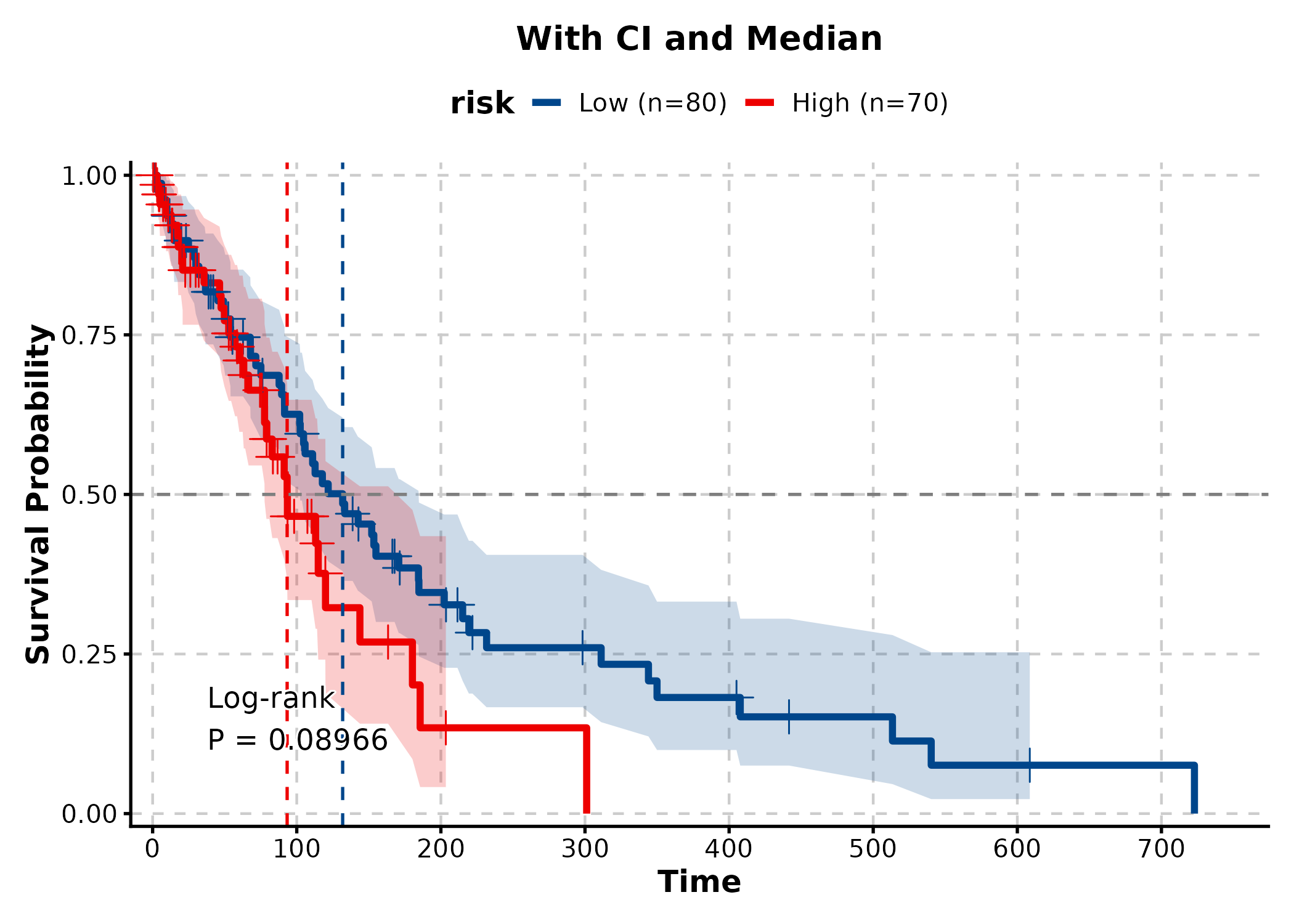
KMPlot(surv, time = "time", status = "status", group_by = "risk",
palette = "nejm", show_risk_table = TRUE, show_pval = TRUE,
show_conf_int = TRUE, title = "Publication-Ready KM")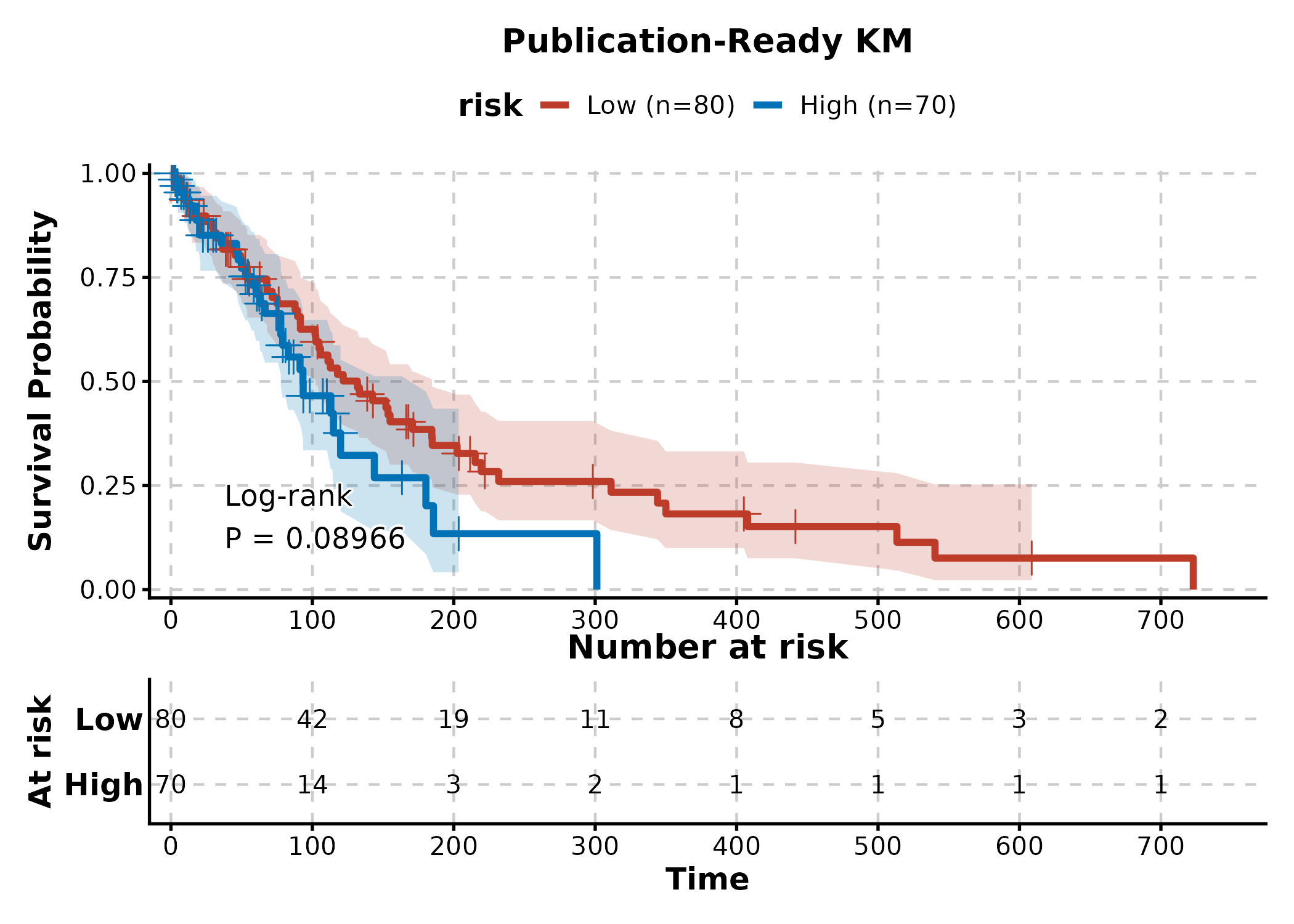
CoxPlot
cox_df <- data.frame(
time = rexp(200, 0.01),
event = sample(0:1, 200, replace = TRUE, prob = c(0.3, 0.7)),
age = rnorm(200, 60, 10),
gender = sample(c("Male", "Female"), 200, replace = TRUE),
stage = sample(c("I-II", "III-IV"), 200, replace = TRUE)
)
CoxPlot(cox_df, time = "time", event = "event",
vars = c("age", "gender", "stage"),
plot_type = "forest", palette = "nejm",
title = "Cox Forest Plot")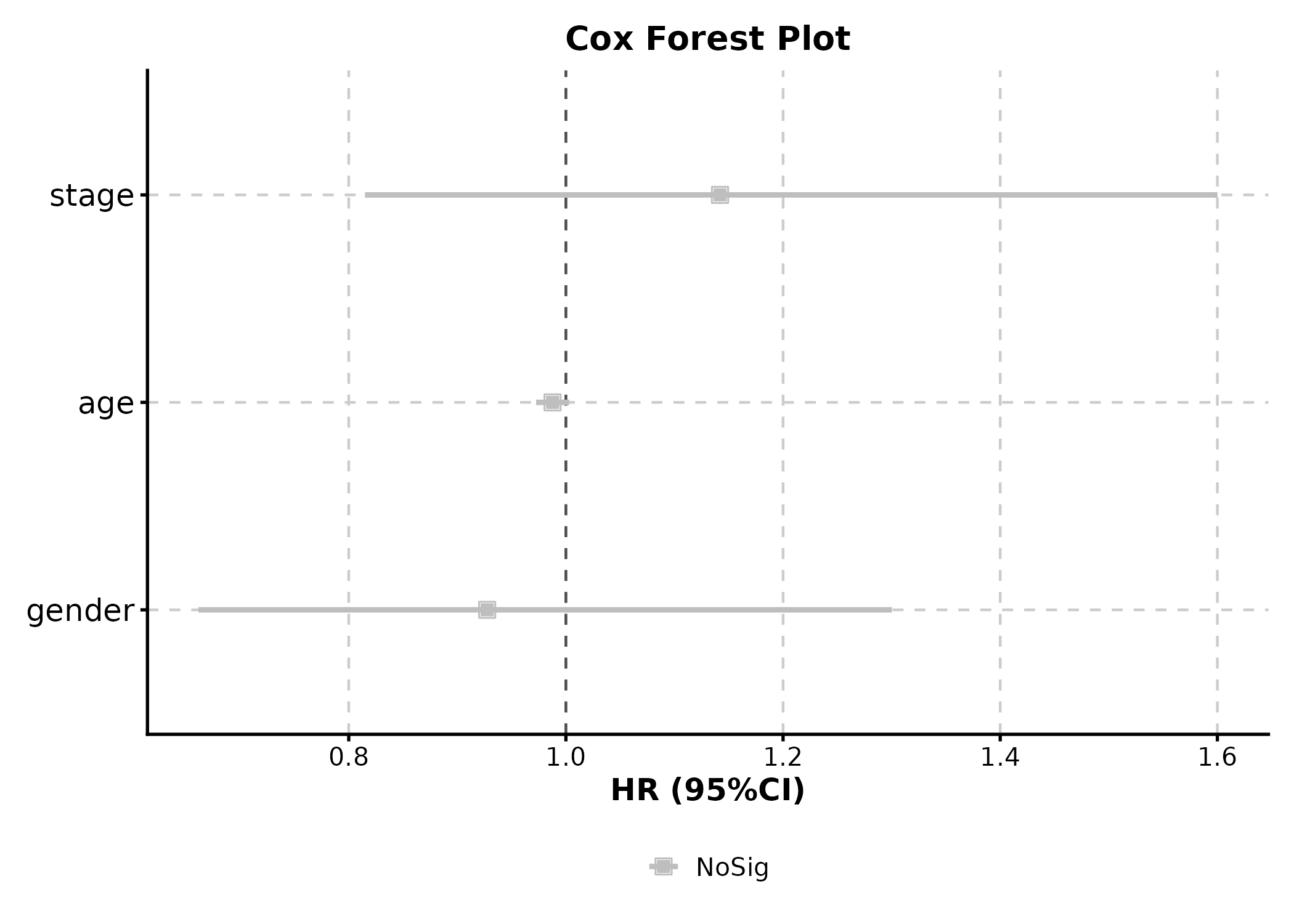
CoxPlot(cox_df, time = "time", event = "event",
vars = c("age", "gender", "stage"),
plot_type = "forest2", palette = "lancet", digits = 2,
title = "Detailed Forest with Stats Table")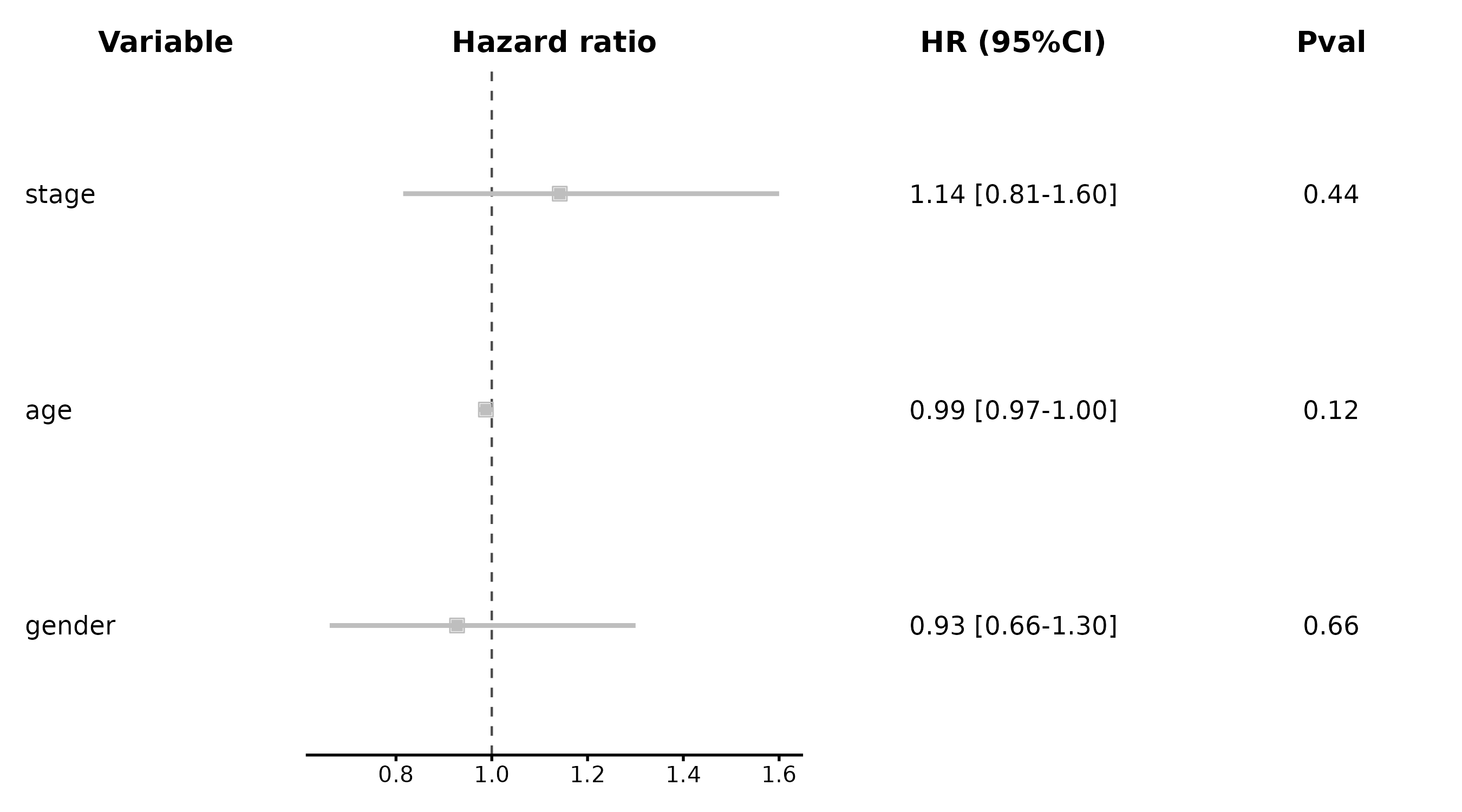
ROCCurve
roc_df <- data.frame(
truth = sample(0:1, 200, replace = TRUE),
score = rnorm(200)
)
roc_df$score <- roc_df$score + roc_df$truth * 1.5
ROCCurve(roc_df, truth_by = "truth", score_by = "score",
palette = "lancet", title = "ROC Analysis")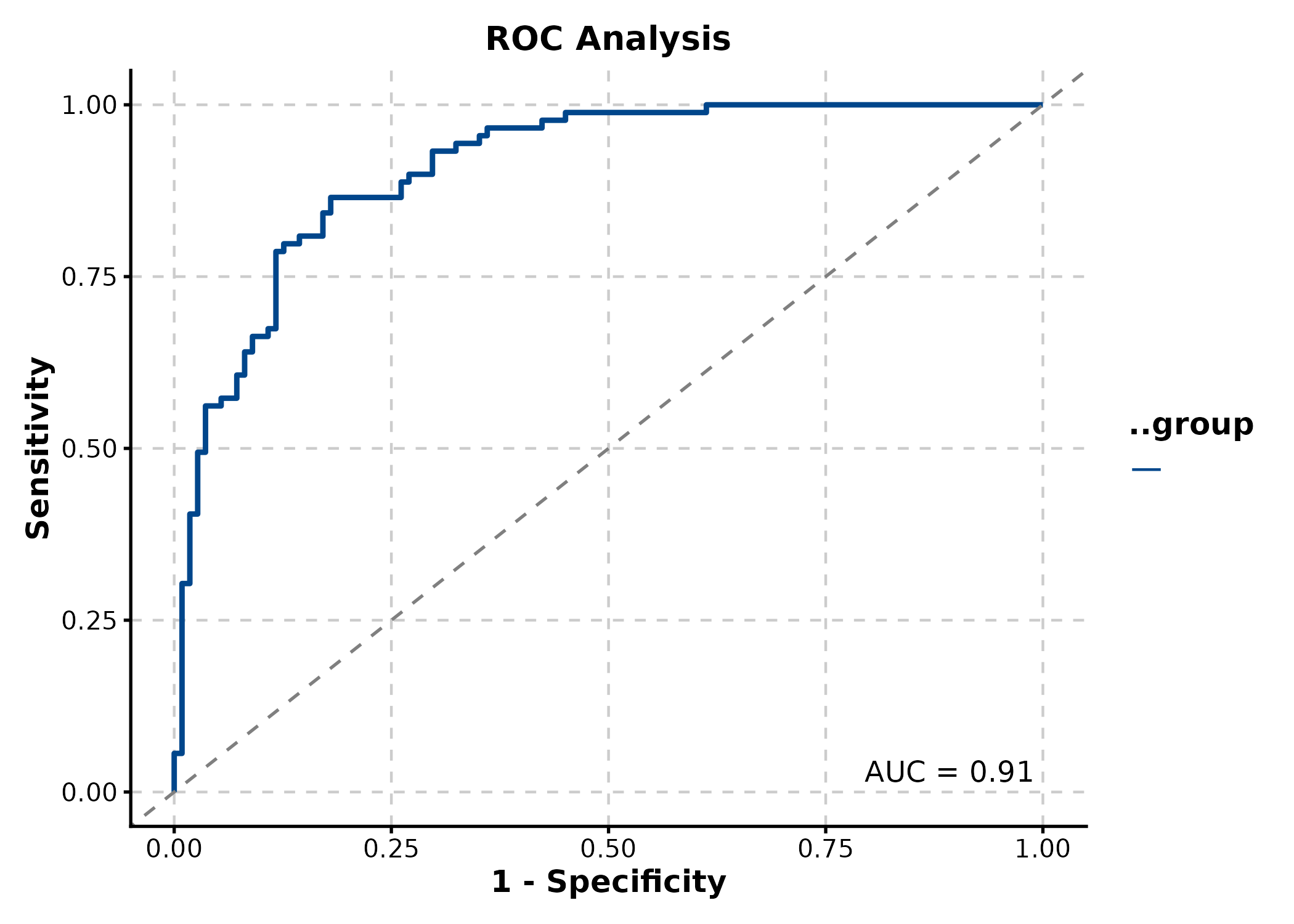
CalibrationPlot
predicted = predicted probabilities,
observed = binary outcome.
cal_df <- data.frame(
outcome = sample(0:1, 500, replace = TRUE),
pred = runif(500)
)
cal_df$pred <- ifelse(cal_df$outcome == 1,
pmin(cal_df$pred + 0.2, 1), pmax(cal_df$pred - 0.2, 0))
CalibrationPlot(cal_df, predicted = "pred", observed = "outcome",
title = "Calibration (LOESS)")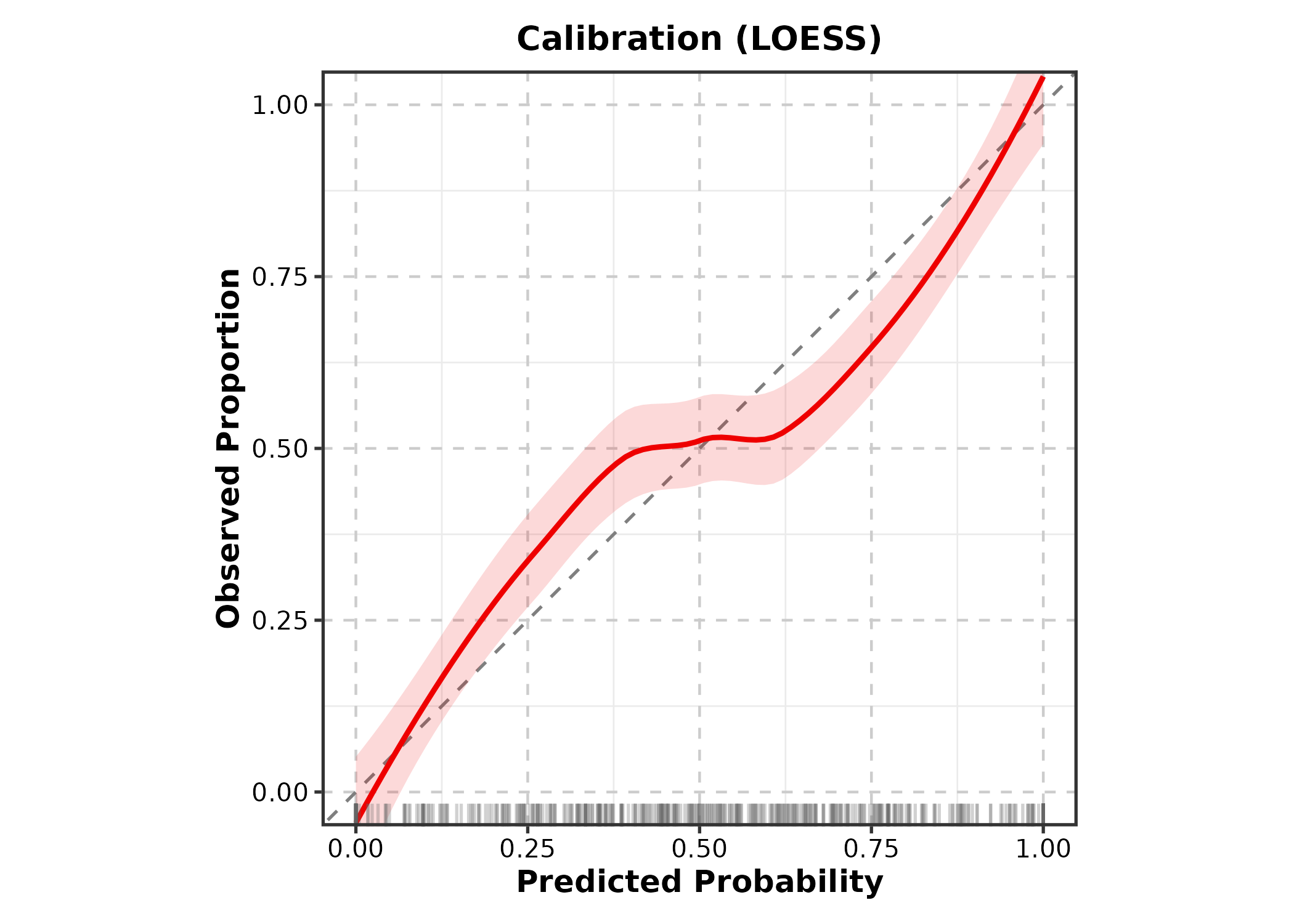
CalibrationPlot(cal_df, predicted = "pred", observed = "outcome",
method = "bins", title = "Calibration (Bins)")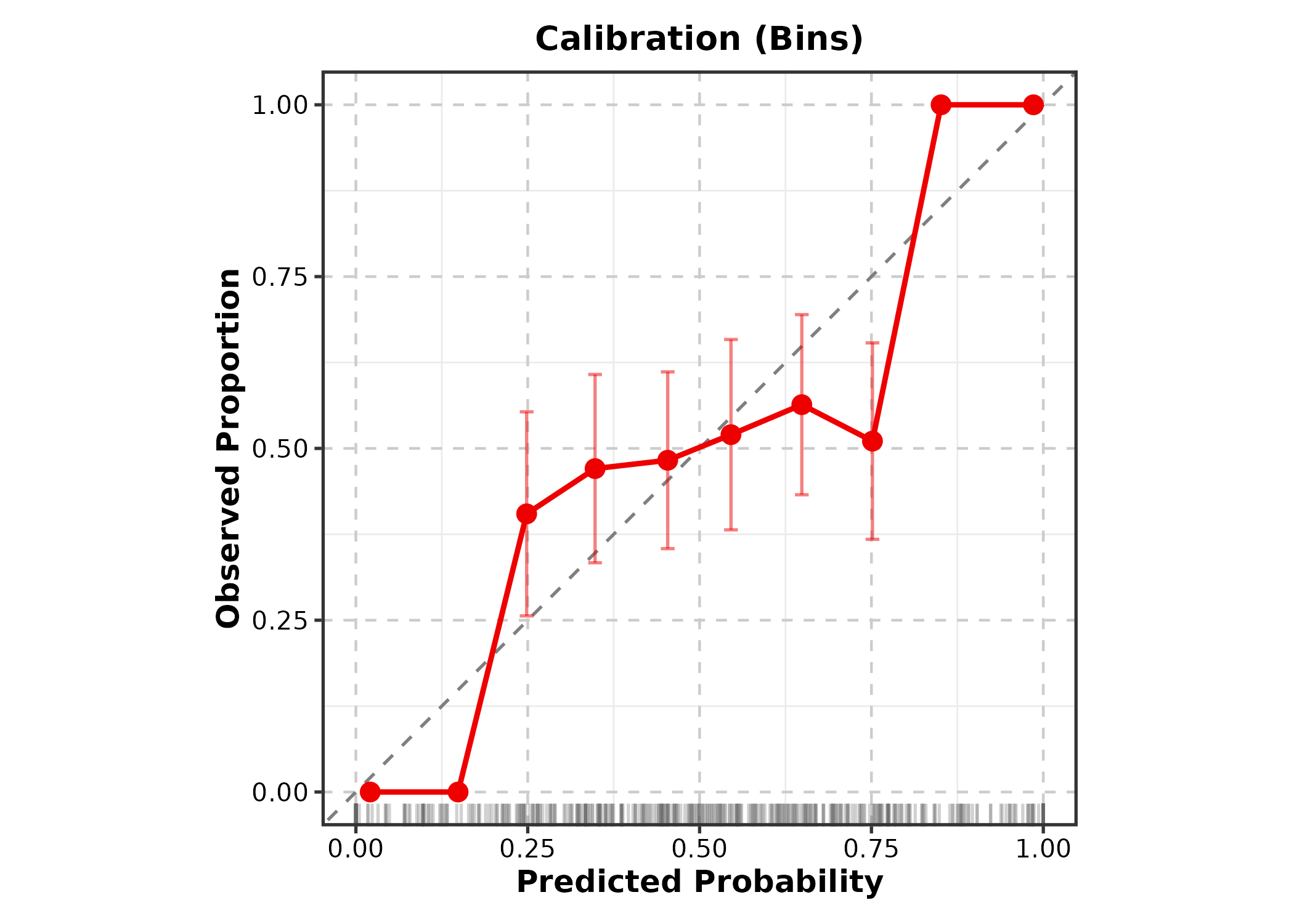
DecisionCurvePlot
outcome = binary outcome column, predictors
= predicted probability column(s).
DecisionCurvePlot(cal_df, outcome = "outcome", predictors = "pred",
title = "Decision Curve Analysis")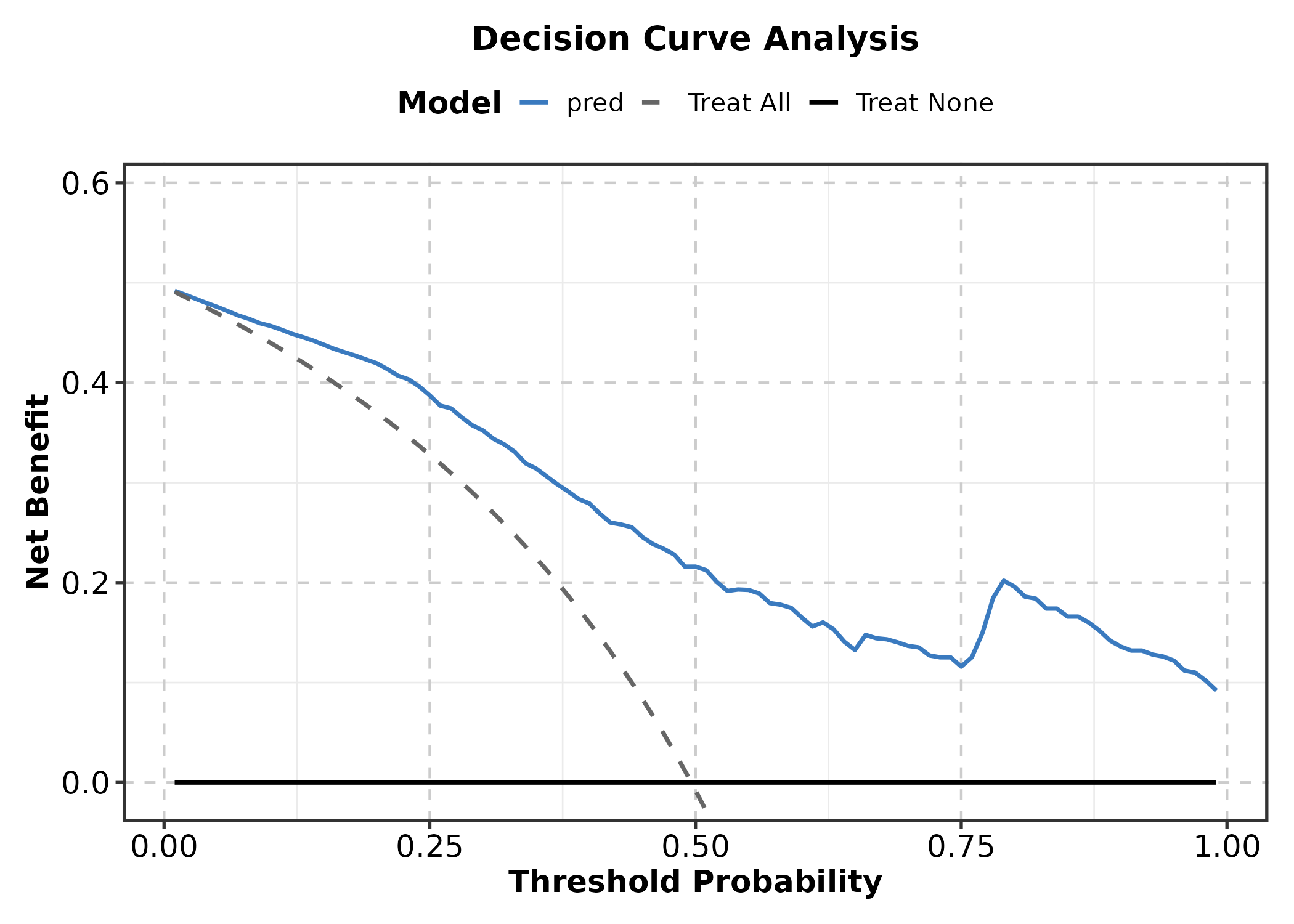
6. Networks & Relationships
Heatmap
genes <- c("CD3D","CD3E","CD8A","FOXP3","CD19","MS4A1",
"CD68","CSF1R","NCAM1","NKG7")
mat <- matrix(rnorm(120, 0, 1.5), nrow = 10,
dimnames = list(genes, paste0("S", 1:12)))
mat[1:4, 1:4] <- mat[1:4, 1:4] + 2
mat[5:6, 5:8] <- mat[5:6, 5:8] + 2
mat[9:10, 9:12] <- mat[9:10, 9:12] + 2
Heatmap(mat, palette = "RdBu",
show_row_names = TRUE, show_column_names = TRUE,
title = "Basic Heatmap")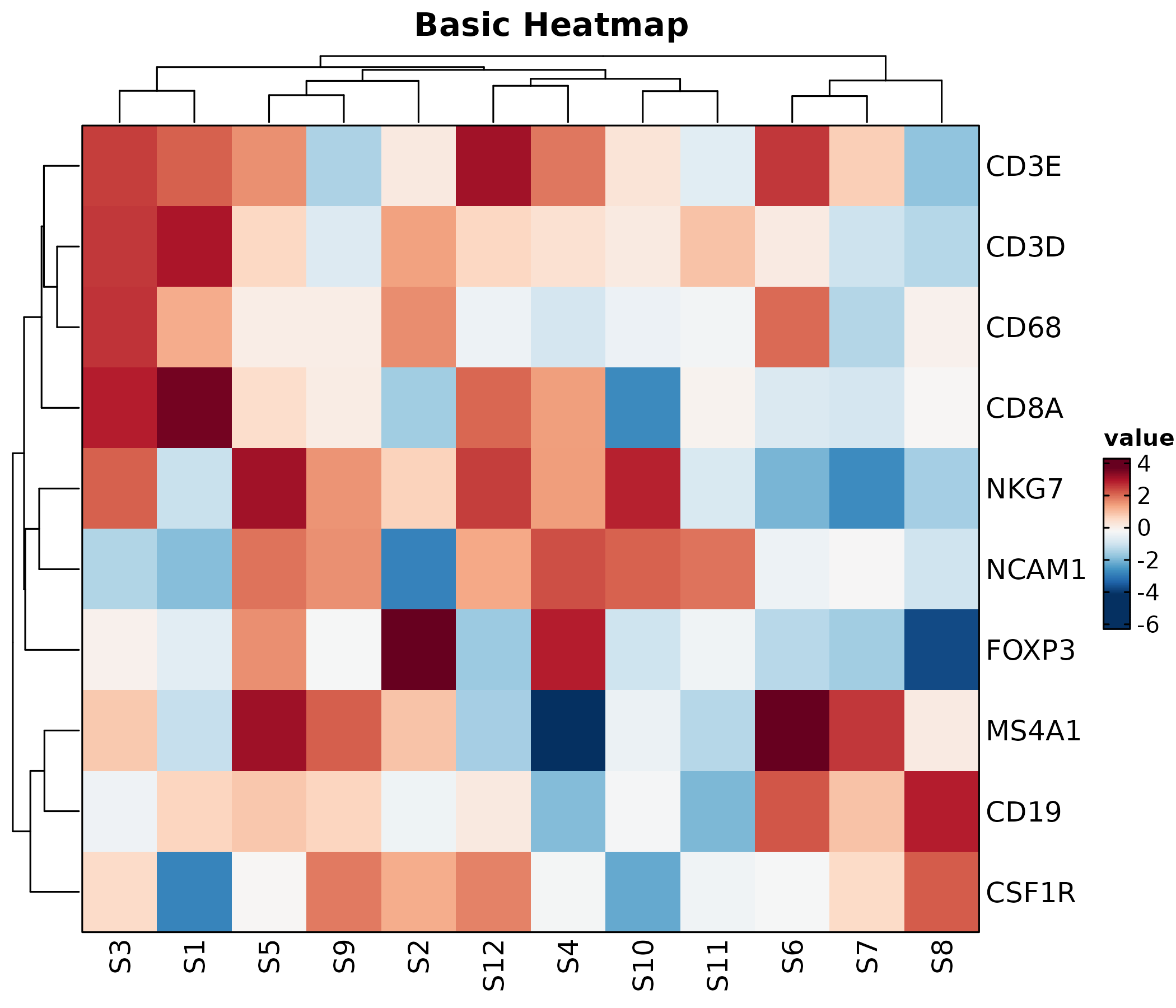
Heatmap(mat, palette = "viridis", values_by = "z-score",
show_row_names = TRUE, show_column_names = TRUE,
rows_name = "Genes", columns_name = "Samples",
title = "Z-score Normalized")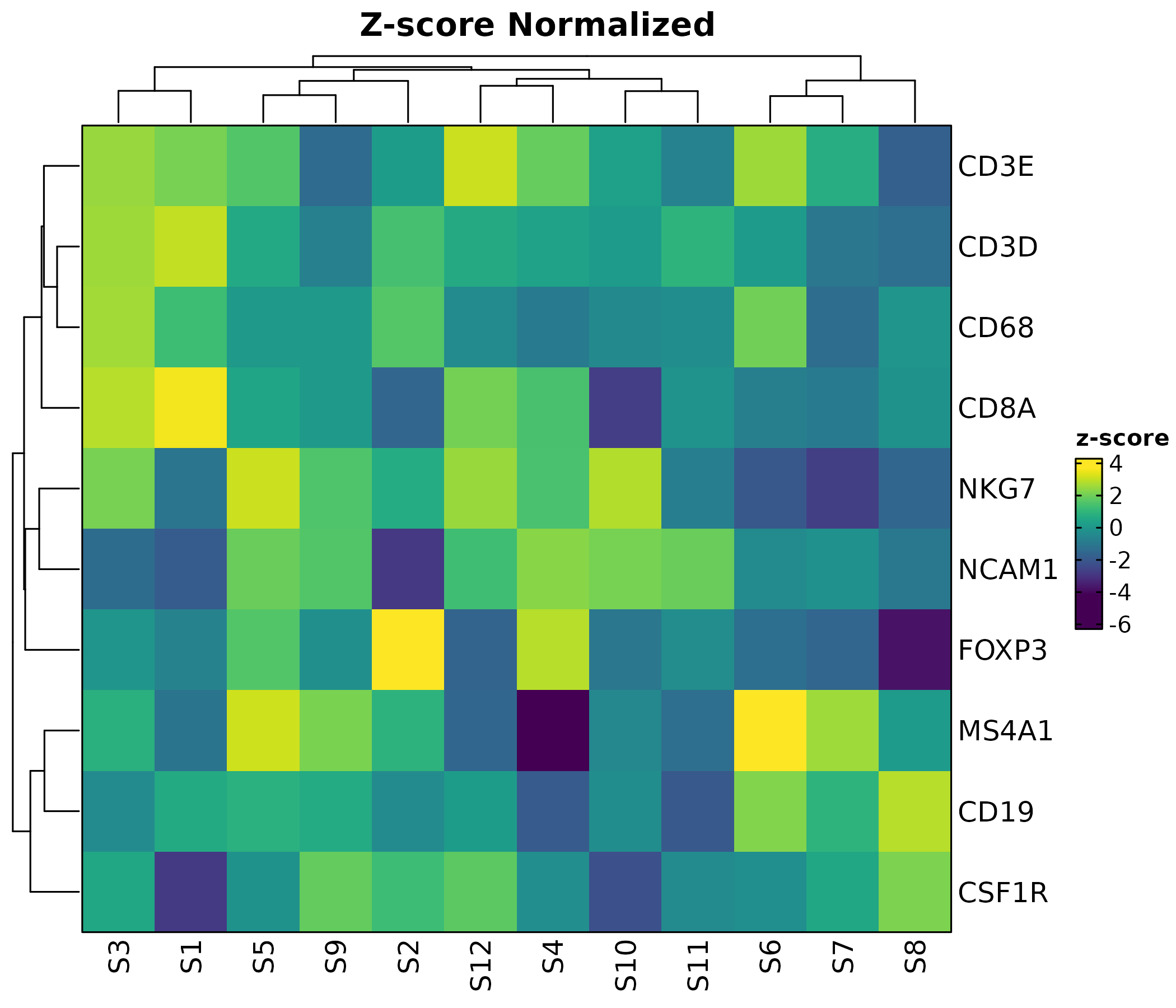
rows_data <- data.frame(
rows = genes,
module = c(rep("T cell", 4), rep("B cell", 2),
rep("Myeloid", 2), rep("NK", 2))
)
Heatmap(mat, palette = "RdBu",
rows_data = rows_data,
rows_annotation = list(Module = "module"),
rows_annotation_palette = list(Module = "Set2"),
show_row_names = TRUE, show_column_names = TRUE,
title = "With Row Annotation")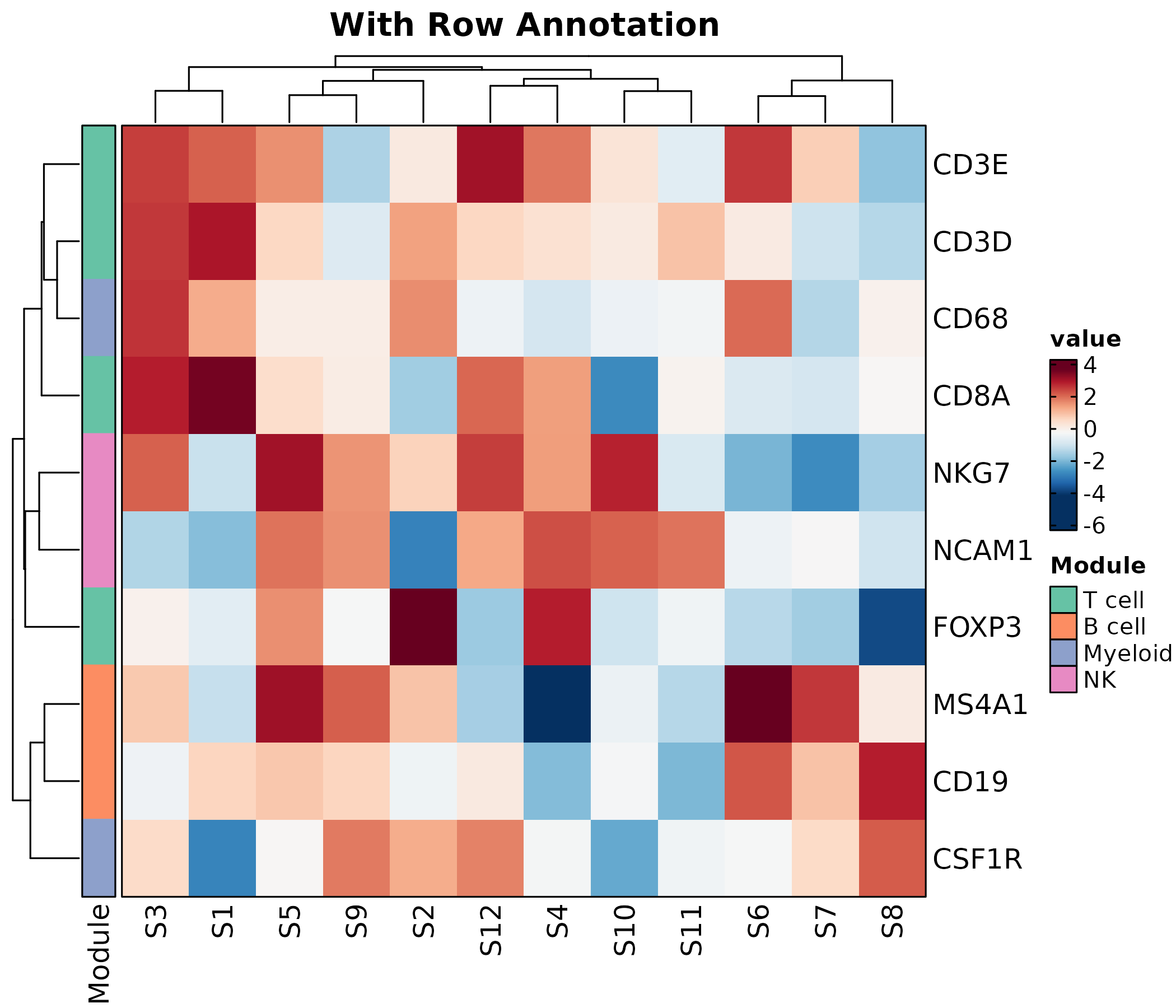
ChordPlot
interactions <- data.frame(
from = c("CD4 T", "CD8 T", "B cell", "NK", "Monocyte"),
to = c("Fibroblast", "Endothelial", "Fibroblast", "Tumor", "Tumor"),
weight = c(15, 20, 10, 25, 18)
)
ChordPlot(interactions, from = "from", to = "to", y = "weight",
palette = "Set3", title = "Chord Diagram")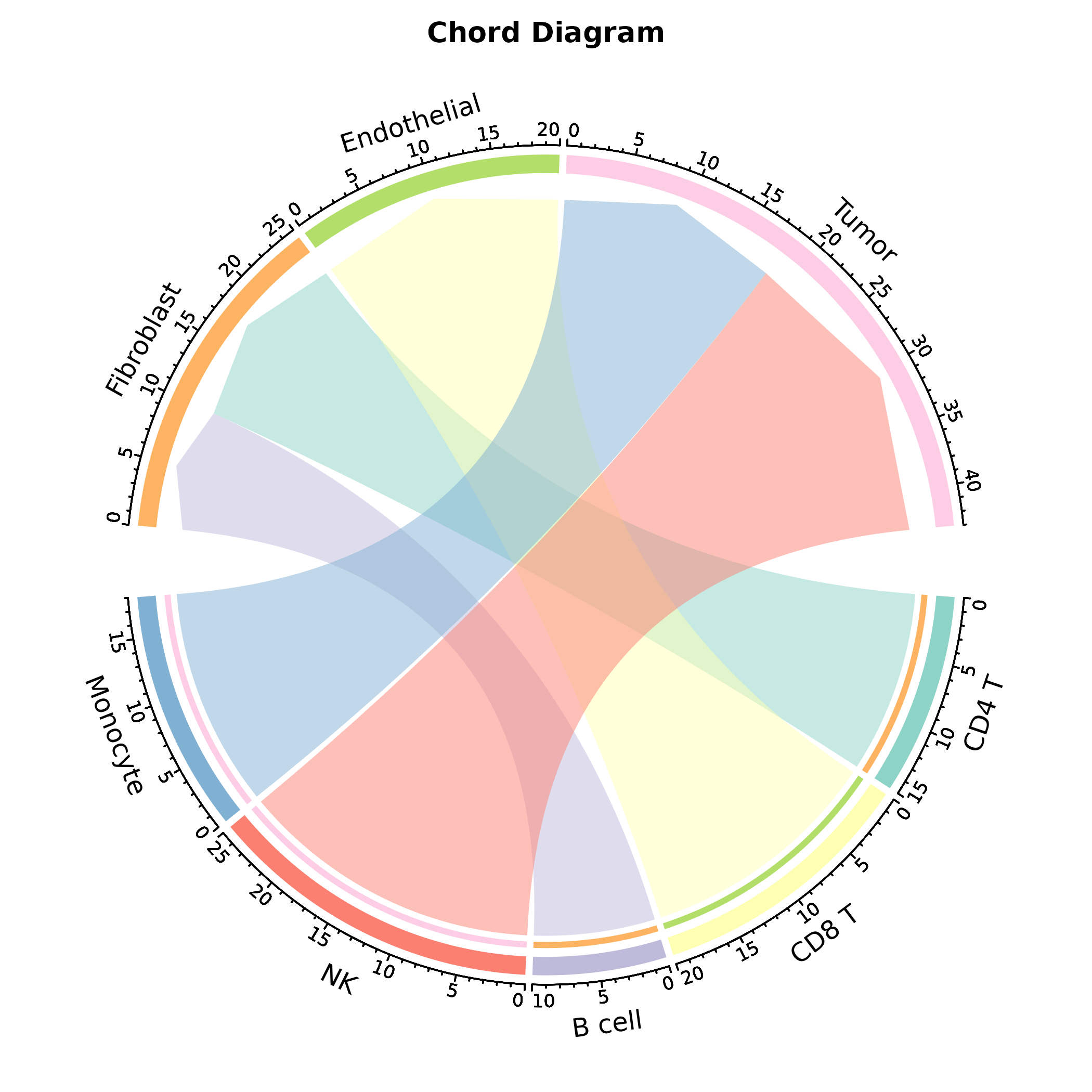
ChordPlot(interactions, from = "from", to = "to", y = "weight",
links_color = "to", labels_rot = TRUE,
palette = "Paired", title = "Colored by Target")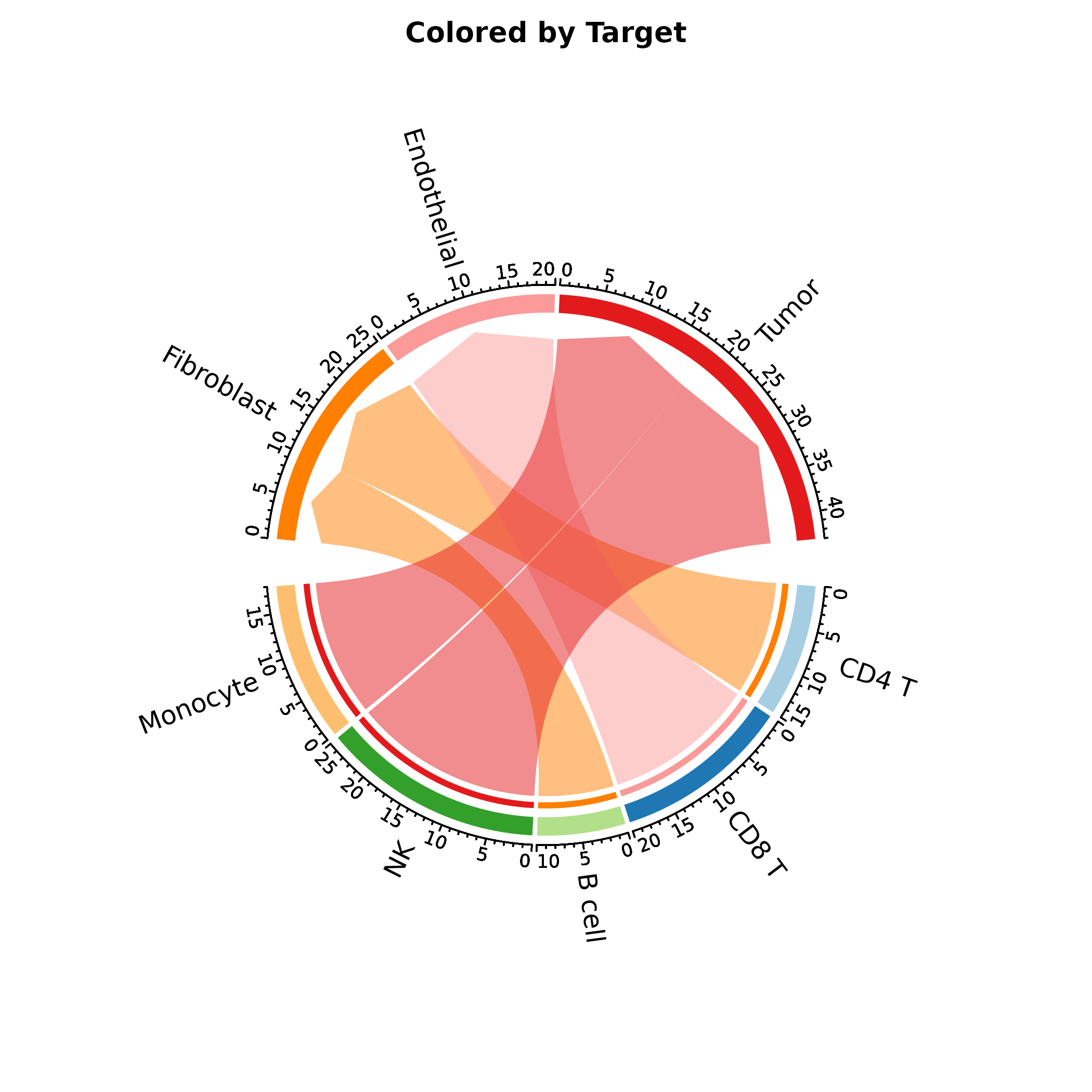
CircosPlot
Alias for ChordPlot.
CircosPlot(interactions, from = "from", to = "to", y = "weight",
palette = "Set3", title = "Circos Plot")SankeyPlot
flow <- data.frame(
diagnosis = rep(c("Early", "Advanced", "Metastatic"), each = 3),
outcome = rep(c("CR", "PR", "PD"), 3),
n = c(40, 8, 2, 10, 20, 10, 2, 8, 20)
)
SankeyPlot(flow, x = c("diagnosis", "outcome"), y = "n",
links_fill_by = "diagnosis", palette = "Set3",
title = "Treatment Flow")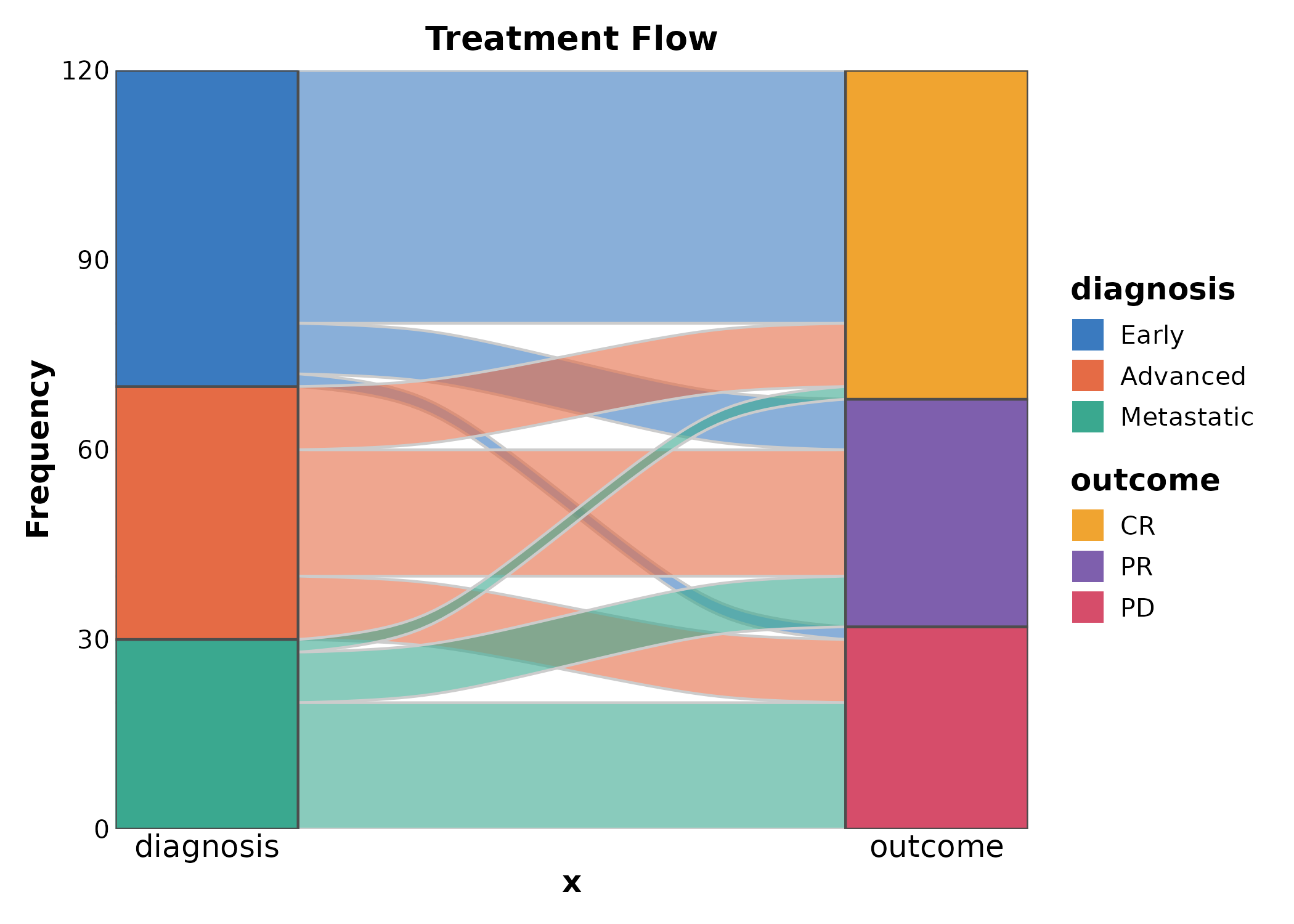
SankeyPlot(flow, x = c("diagnosis", "outcome"), y = "n",
links_fill_by = "outcome", palette = "npg",
title = "Colored by Outcome")
AlluvialPlot
Alias for SankeyPlot.
AlluvialPlot(flow, x = c("diagnosis", "outcome"), y = "n",
links_fill_by = "diagnosis", palette = "Paired",
title = "Alluvial Plot")
Network
edges <- data.frame(
from = c("TP53", "EGFR", "MYC", "KRAS", "BRCA1", "PTEN"),
to = c("MDM2", "ERBB2", "MAX", "BRAF", "RAD51", "AKT1"),
score = c(0.9, 0.85, 0.8, 0.75, 0.7, 0.65)
)
nodes <- data.frame(
name = unique(c(edges$from, edges$to)),
type = c("Oncogene","Oncogene","Oncogene","Oncogene",
"TSG","TSG","TSG","Oncogene","Oncogene",
"Oncogene","TSG","Oncogene")
)
Network(edges, nodes, link_weight_by = "score",
node_fill_by = "type", layout = "fr",
title = "Protein Interaction Network")
7. Specialized
DumbbellPlot
x_start and x_end are the numeric value
columns, y is the category.
dumb <- data.frame(
gene = paste0("Gene", LETTERS[1:8]),
before = rnorm(8, 5, 1.5), after = rnorm(8, 8, 1.5)
)
DumbbellPlot(dumb, x_start = "before", x_end = "after", y = "gene",
palette = "Set1", title = "Before vs After Treatment")
DumbbellPlot(dumb, x_start = "before", x_end = "after", y = "gene",
palette = "lancet", sort_y = "desc",
title = "Sorted by Difference")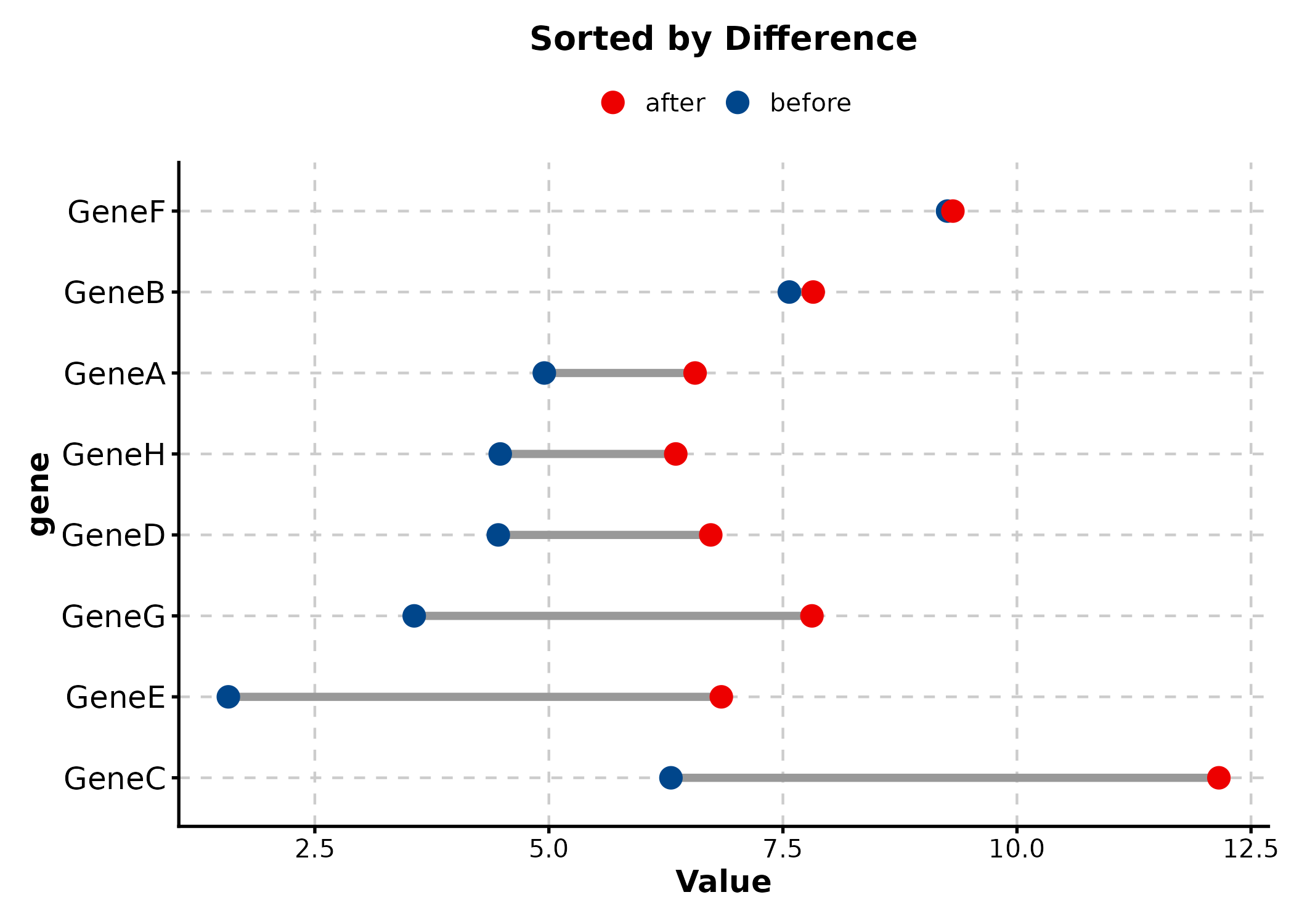
StreamGraph
stream_df <- data.frame(
year = rep(2015:2024, 4),
count = abs(rnorm(40, 50, 20)),
category = rep(c("Immunotherapy", "Targeted", "Chemo", "Radiation"), each = 10)
)
StreamGraph(stream_df, x = "year", y = "count", group_by = "category",
palette = "Set2", title = "Treatment Trends")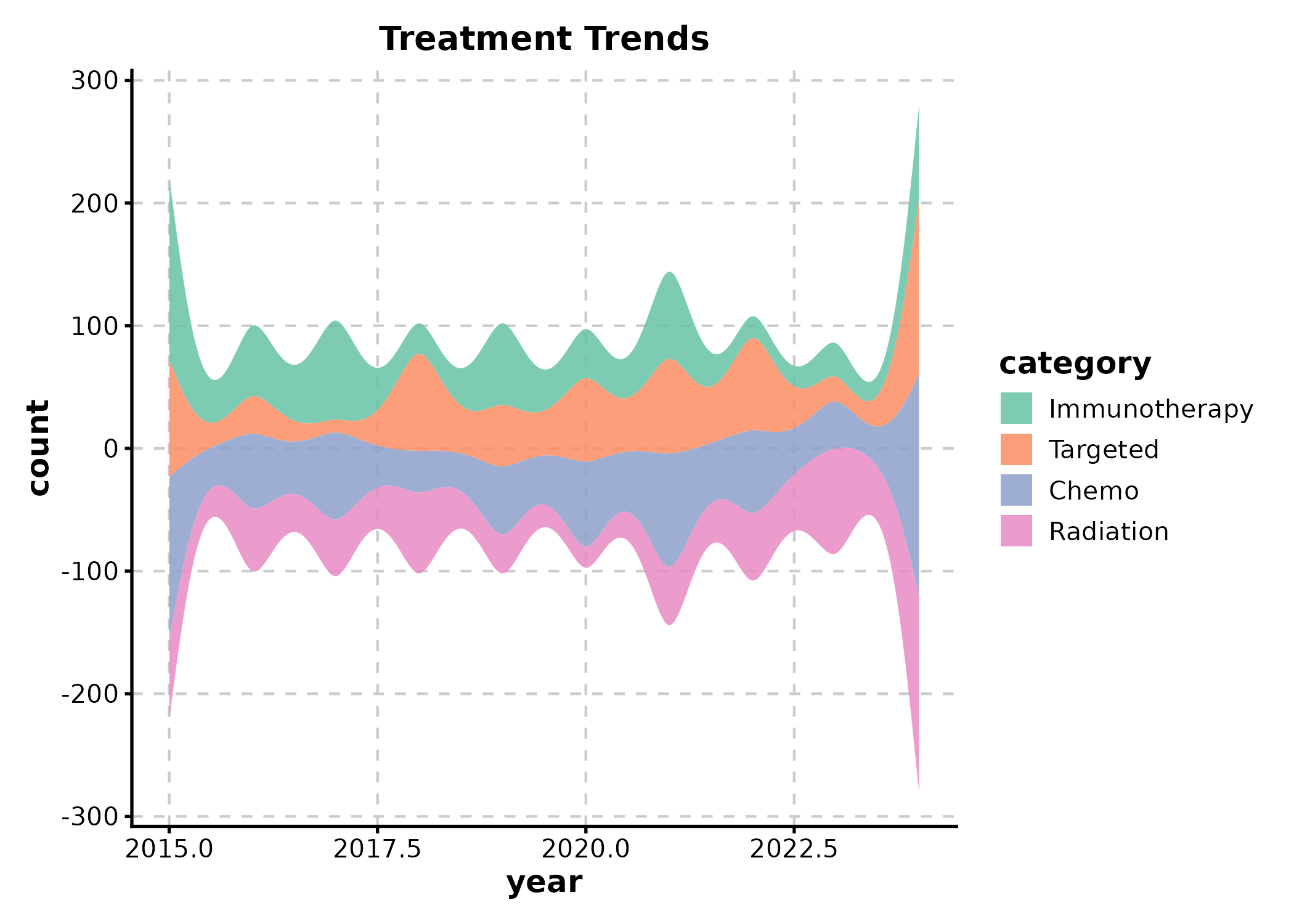
TreemapPlot
area = numeric size column, label = text
label, fill = fill group.
treemap_df <- data.frame(
pathway = c("PI3K-Akt", "MAPK", "Wnt", "p53", "Cell Cycle",
"Apoptosis", "JAK-STAT", "NF-kB"),
gene_count = c(45, 38, 32, 28, 25, 22, 18, 15),
category = c("Signaling","Signaling","Signaling","TSG",
"Cell Cycle","Death","Signaling","Signaling")
)
TreemapPlot(treemap_df, area = "gene_count", label = "pathway",
fill = "category", palette = "Set2",
title = "Pathway Gene Counts")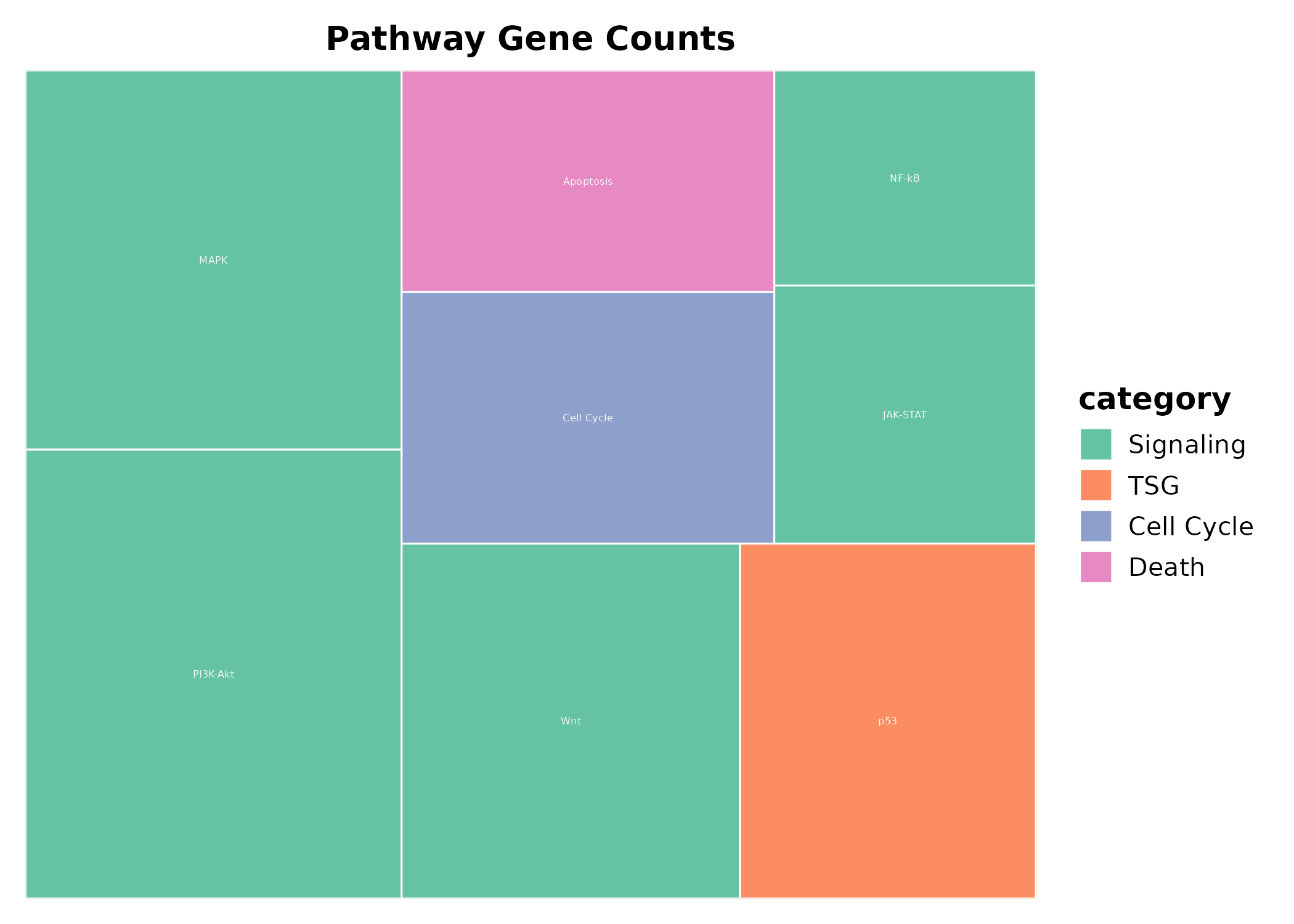
ParallelCoordPlot
ParallelCoordPlot(iris,
columns = c("Sepal.Length", "Sepal.Width", "Petal.Length", "Petal.Width"),
group_by = "Species", palette = "Set1",
title = "Iris Features")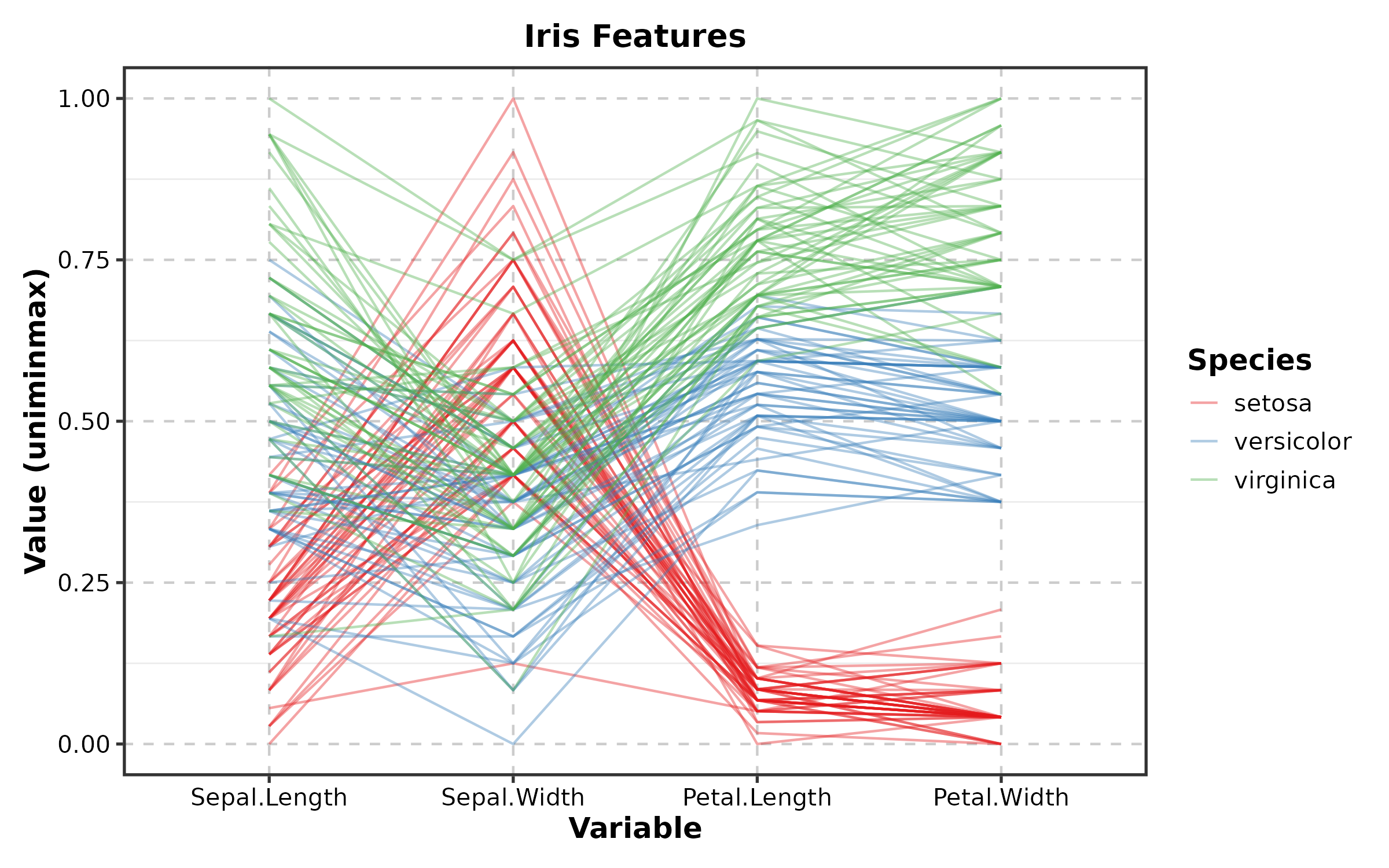
TimelinePlot
events <- data.frame(
event = c("Diagnosis", "Surgery", "Chemo", "Radiation", "Follow-up"),
start = as.Date(c("2024-01-15", "2024-02-10", "2024-03-01",
"2024-06-01", "2024-09-01")),
end = as.Date(c("2024-01-15", "2024-02-12", "2024-05-15",
"2024-07-15", "2024-12-01"))
)
TimelinePlot(events, start = "start", end = "end", label = "event",
palette = "Set2", title = "Treatment Timeline")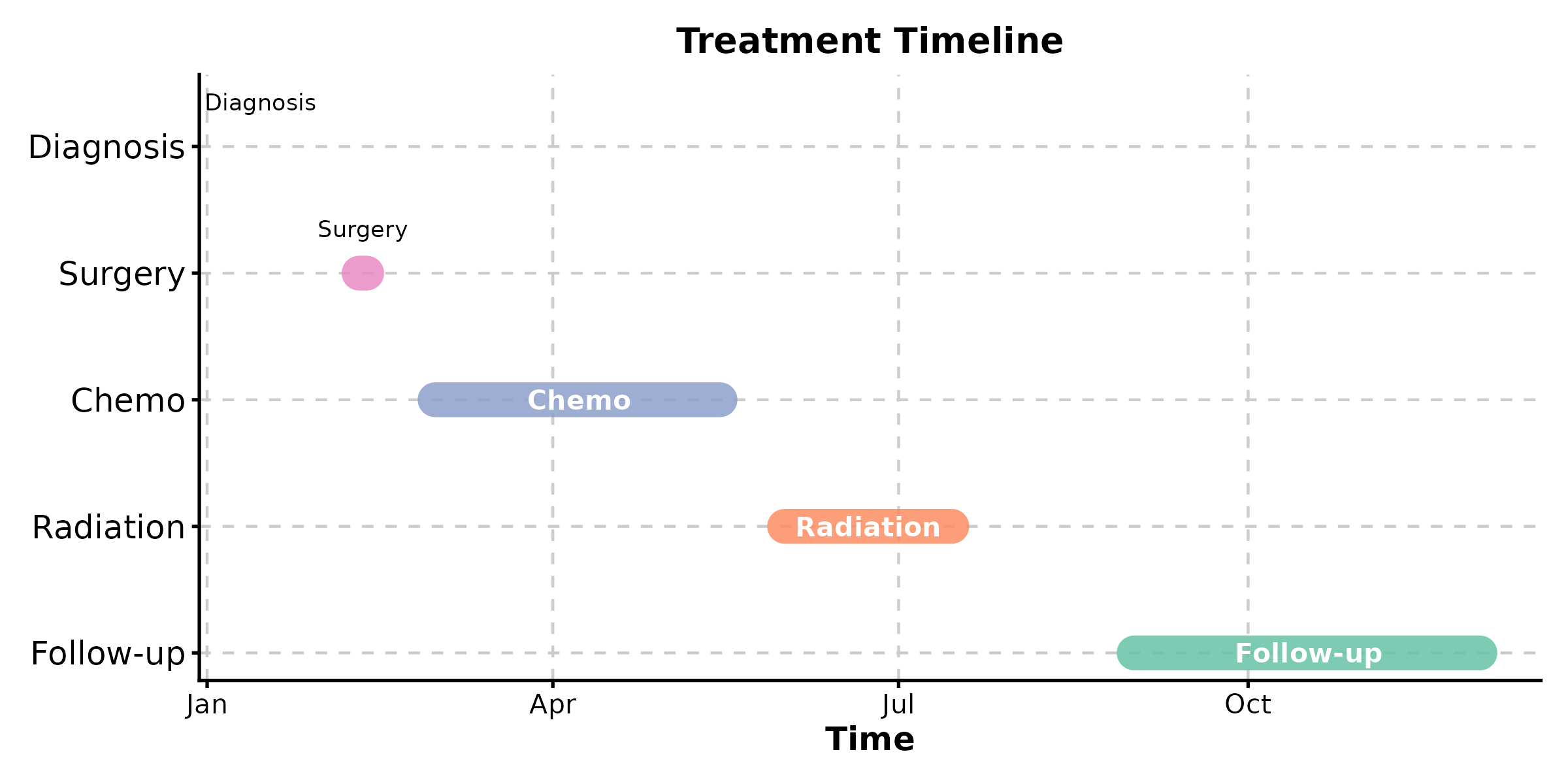
DendrogramPlot
DendrogramPlot(mtcars[1:15, ],
columns = c("mpg", "disp", "hp", "wt", "qsec"),
k = 3, palette = "Set1",
title = "Hierarchical Clustering")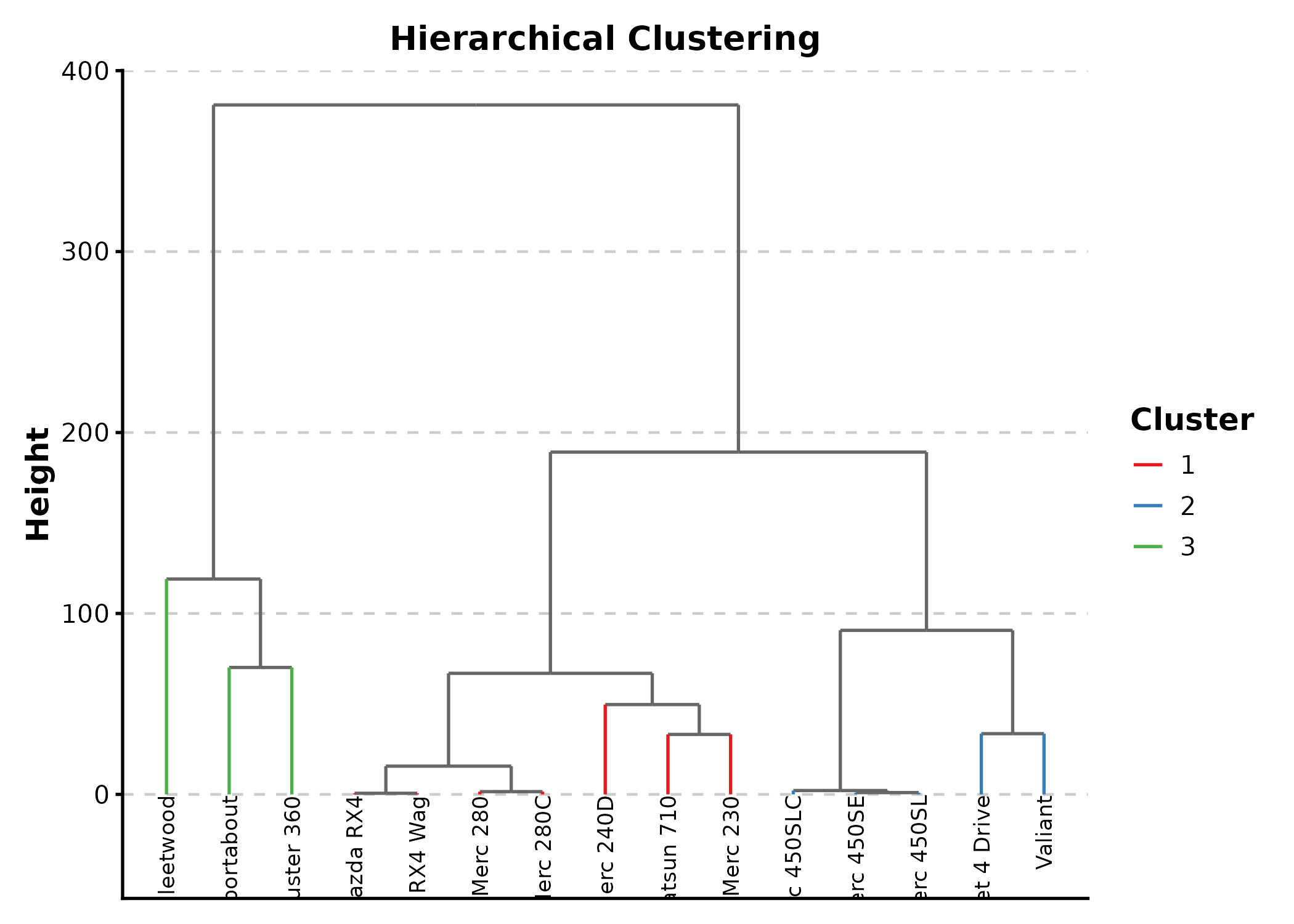
SunburstPlot
Requires plotly. Returns interactive output.
SunburstPlot(sunburst_data,
labels = "label", parents = "parent", values = "count",
title = "Hierarchical Composition")WordCloudPlot
terms <- data.frame(
word = c("apoptosis", "proliferation", "migration", "invasion",
"angiogenesis", "metastasis", "differentiation",
"inflammation", "autophagy", "senescence"),
score = c(8, 7, 6, 5, 9, 4, 7, 8, 3, 4)
)
WordCloudPlot(terms, word_by = "word", score_by = "score",
palette = "Spectral", title = "Biological Processes")
RarefactionPlot
RarefactionPlot(inext_result, palette = "Set1",
title = "Species Rarefaction")8. Earth & Environmental
ContourPlot
grid_data <- expand.grid(
x = seq(-3, 3, length.out = 50),
y = seq(-3, 3, length.out = 50)
)
grid_data$z <- with(grid_data, sin(x) * cos(y) * exp(-(x^2 + y^2) / 8))
ContourPlot(grid_data, x = "x", y = "y", z = "z",
type = "filled", palette = "Spectral",
title = "Filled Contour")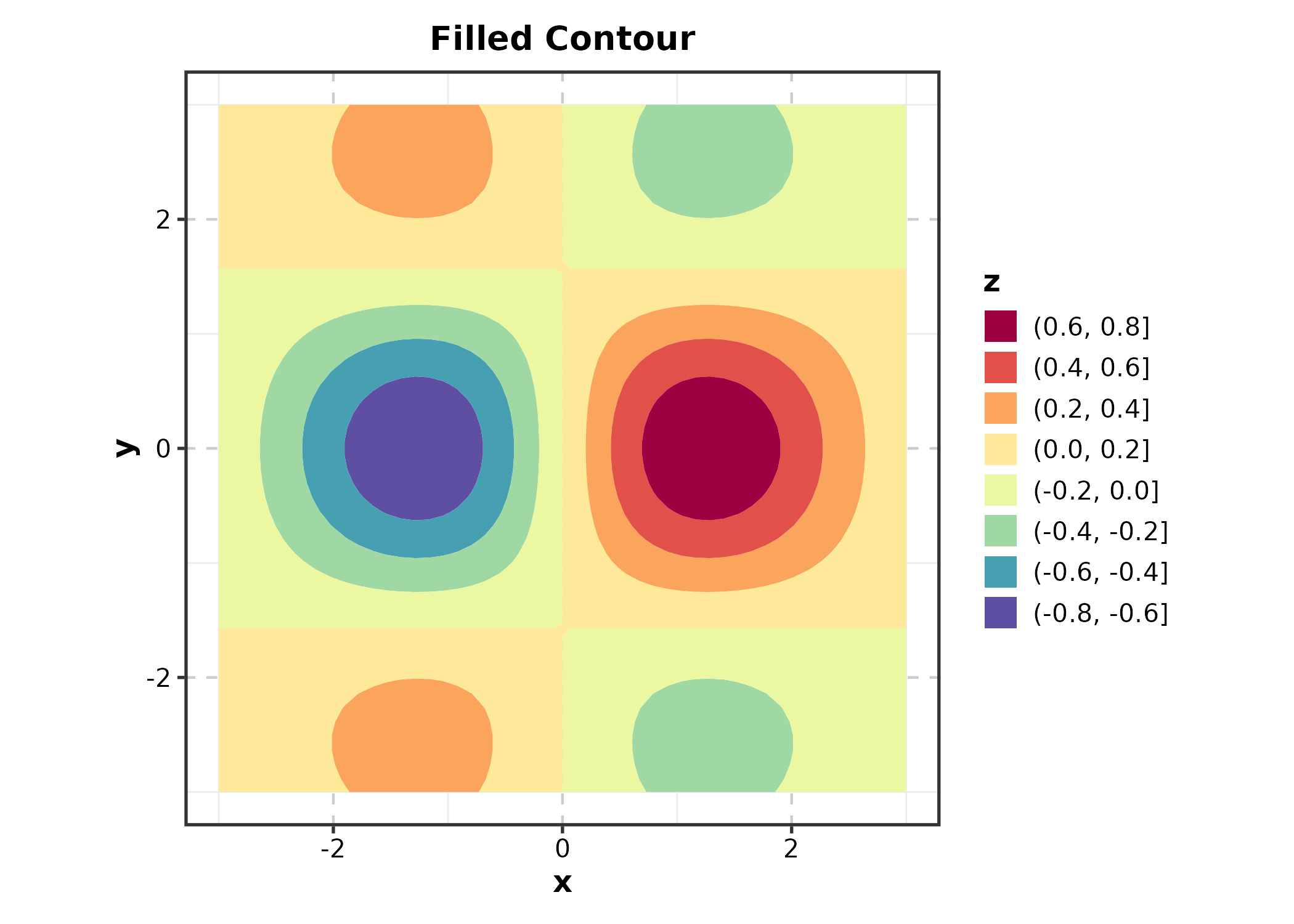
ContourPlot(grid_data, x = "x", y = "y", z = "z",
type = "lines", palette = "viridis",
title = "Contour Lines")
ContourPlot(grid_data, x = "x", y = "y", z = "z",
type = "both", palette = "RdBu",
title = "Lines on Filled")
TernaryPlot
Requires ggtern (namespace may conflict with
ggplot2).
ternary_df <- data.frame(
sand = c(0.4, 0.2, 0.6), silt = c(0.3, 0.5, 0.2),
clay = c(0.3, 0.3, 0.2), type = c("Loam", "Silty", "Sandy")
)
TernaryPlot(ternary_df, x = "sand", y = "silt", z = "clay",
group_by = "type", palette = "Set1",
title = "Soil Composition")PolarPlot
theta = angle variable, r = radius
variable.
wind <- data.frame(
direction = rep(seq(0, 350, by = 10), 3),
speed = abs(rnorm(108, 10, 5)),
season = rep(c("Spring", "Summer", "Autumn"), each = 36)
)
PolarPlot(wind, theta = "direction", r = "speed",
group_by = "season", palette = "Set2",
title = "Wind Pattern")
PolarPlot(wind, theta = "direction", r = "speed",
group_by = "season", palette = "npg", type = "scatter",
title = "Polar Scatter")
WindRosePlot
Alias for PolarPlot.
WindRosePlot(wind, theta = "direction", r = "speed",
group_by = "season", palette = "Set1",
title = "Wind Rose")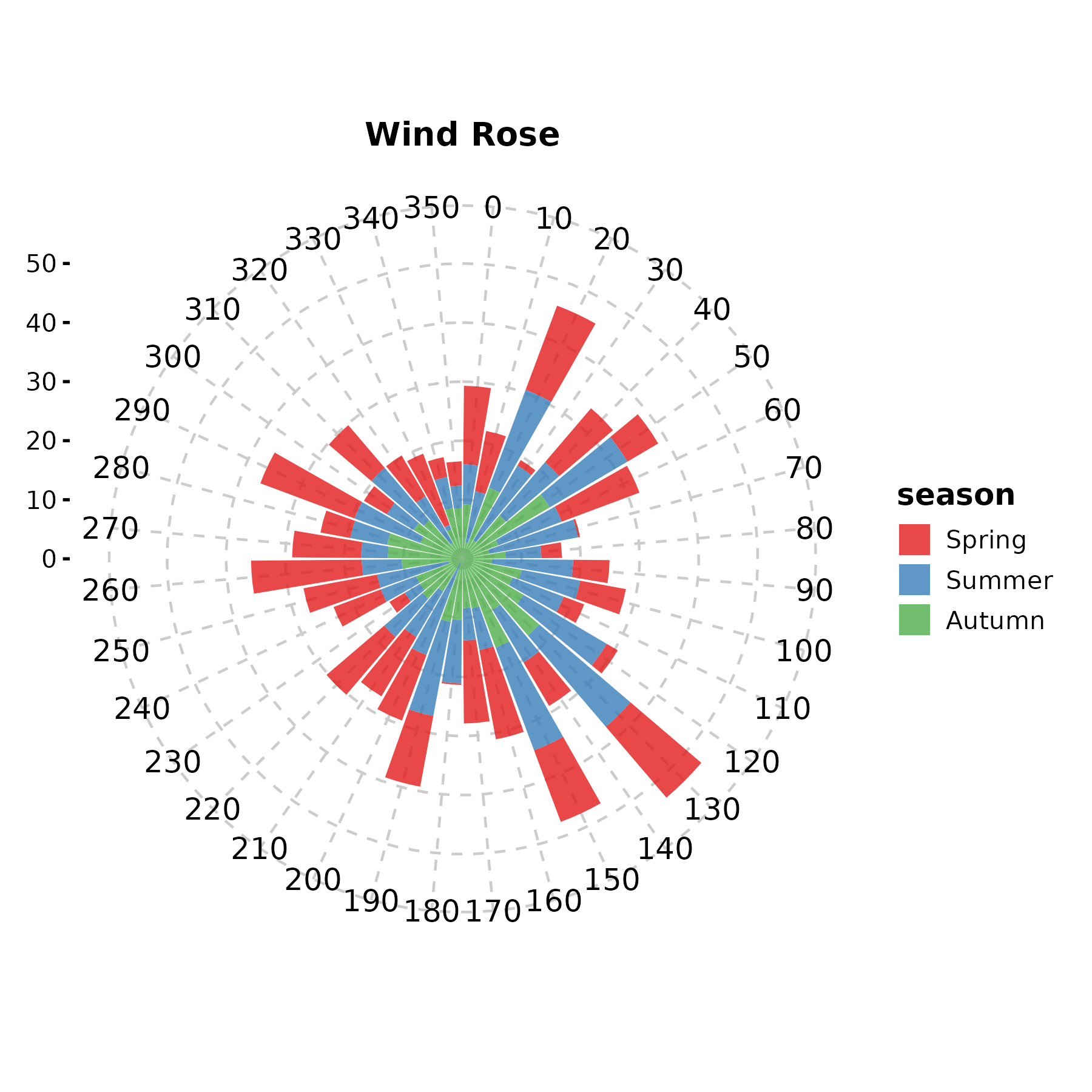
MapPlot
Requires rnaturalearth and
rnaturalearthdata.
cities <- data.frame(
lon = c(-73.9, -87.6, -118.2, -122.4),
lat = c(40.7, 41.9, 34.1, 37.8),
pop = c(8.3, 2.7, 3.9, 0.87)
)
MapPlot(cities, type = "point", point_x = "lon", point_y = "lat",
value = "pop", color_name = "Population (M)")9. Meta-Analysis & Agreement
ForestPlot
estimate, ci_lower, ci_upper
are the effect size columns. is_summary marks diamond
rows.
meta <- data.frame(
study = c("Smith 2018", "Chen 2019", "Garcia 2020",
"Kim 2021", "Patel 2022", "Overall"),
or = c(1.32, 0.85, 1.51, 1.08, 0.92, 1.12),
lower = c(0.95, 0.60, 1.05, 0.78, 0.65, 0.93),
upper = c(1.84, 1.20, 2.17, 1.50, 1.30, 1.35),
weight = c(22, 18, 15, 25, 20, NA),
is_summary = c(FALSE, FALSE, FALSE, FALSE, FALSE, TRUE)
)
ForestPlot(meta, estimate = "or", ci_lower = "lower", ci_upper = "upper",
label = "study", weight = "weight", is_summary = "is_summary",
null_value = 1, log_scale = TRUE,
title = "Meta-Analysis Forest Plot")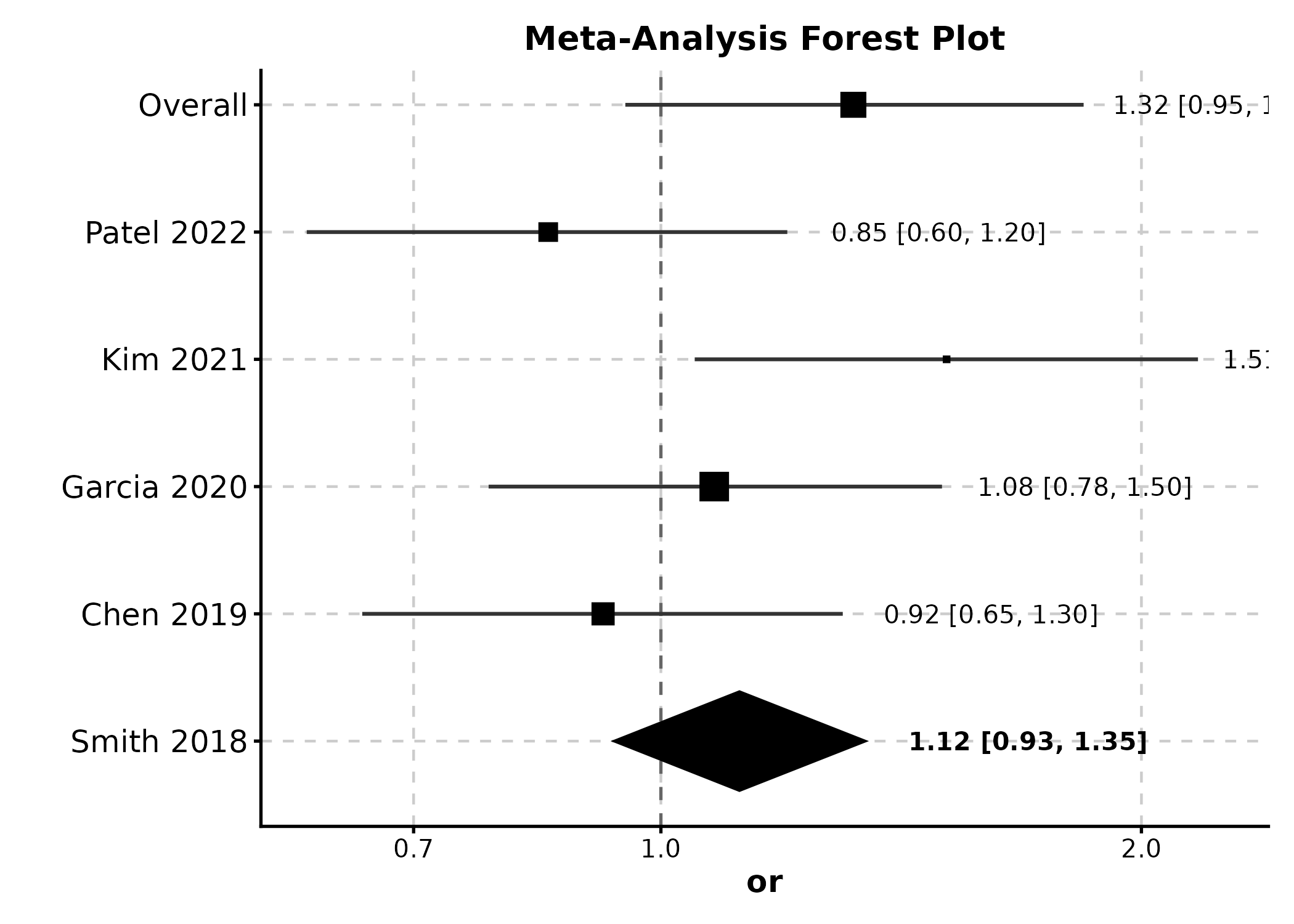
ForestPlot(meta, estimate = "or", ci_lower = "lower", ci_upper = "upper",
label = "study", weight = "weight", is_summary = "is_summary",
null_value = 1, show_values = TRUE,
title = "With Value Labels")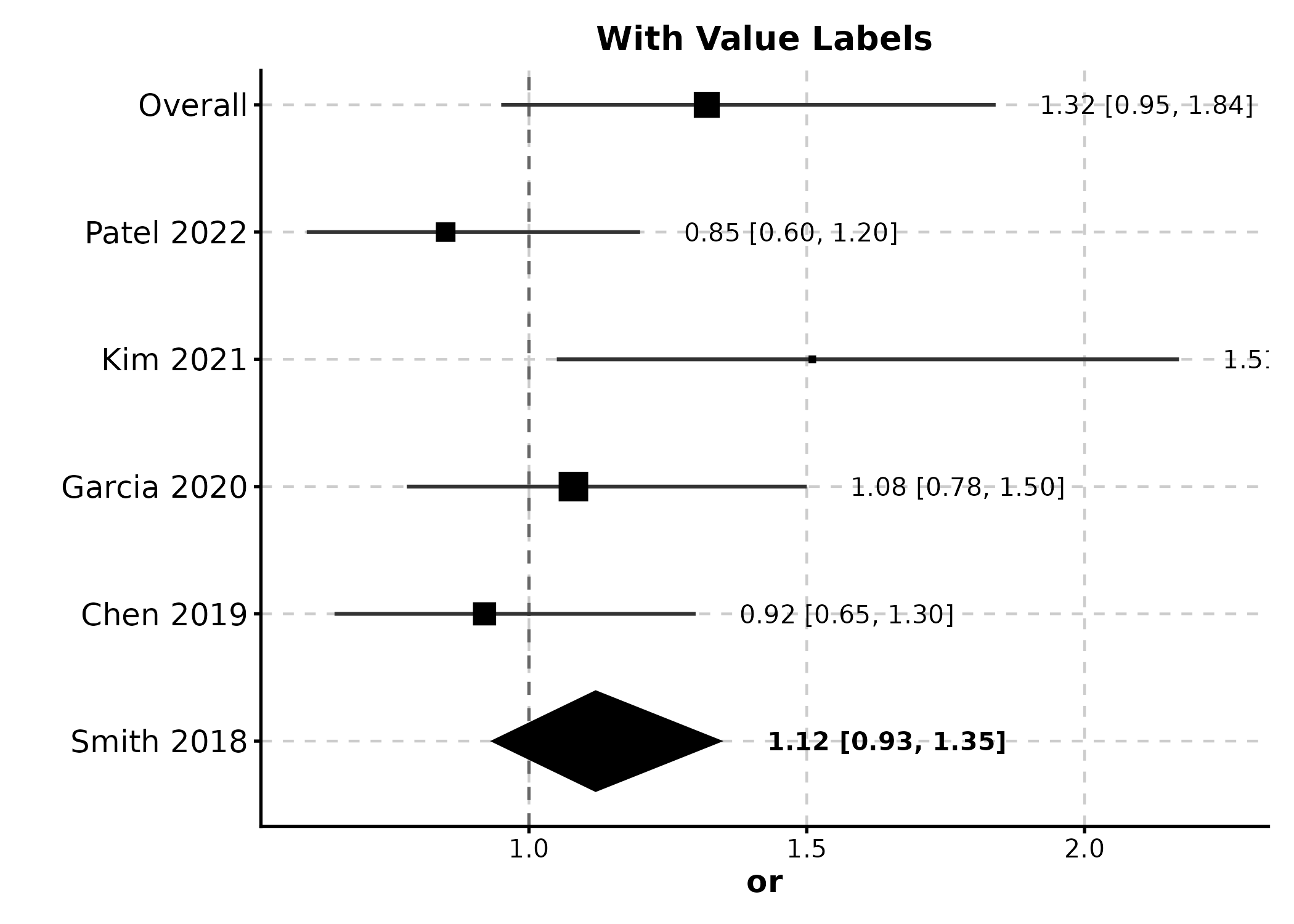
FunnelPlot
estimate = effect sizes, se = standard
errors.
funnel_df <- data.frame(
effect = rnorm(20, 0.5, 0.3),
se = runif(20, 0.05, 0.4)
)
FunnelPlot(funnel_df, estimate = "effect", se = "se",
title = "Publication Bias Assessment")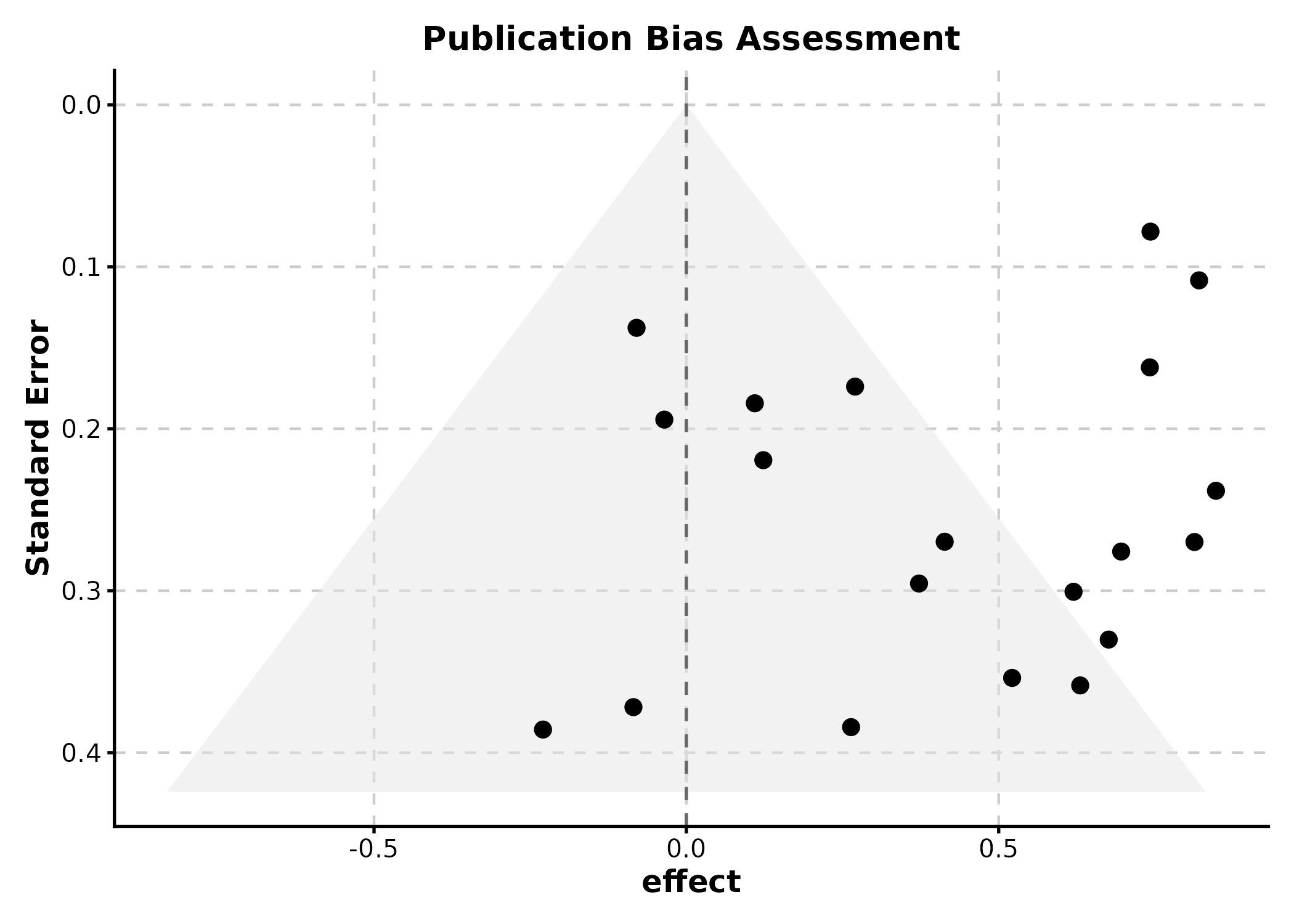
BlandAltmanPlot
method1 and method2 are the two measurement
columns.
ba_df <- data.frame(
method_A = rnorm(50, 100, 15),
method_B = rnorm(50, 100, 15)
)
ba_df$method_B <- ba_df$method_A + rnorm(50, 2, 5)
BlandAltmanPlot(ba_df, method1 = "method_A", method2 = "method_B",
title = "Method Agreement")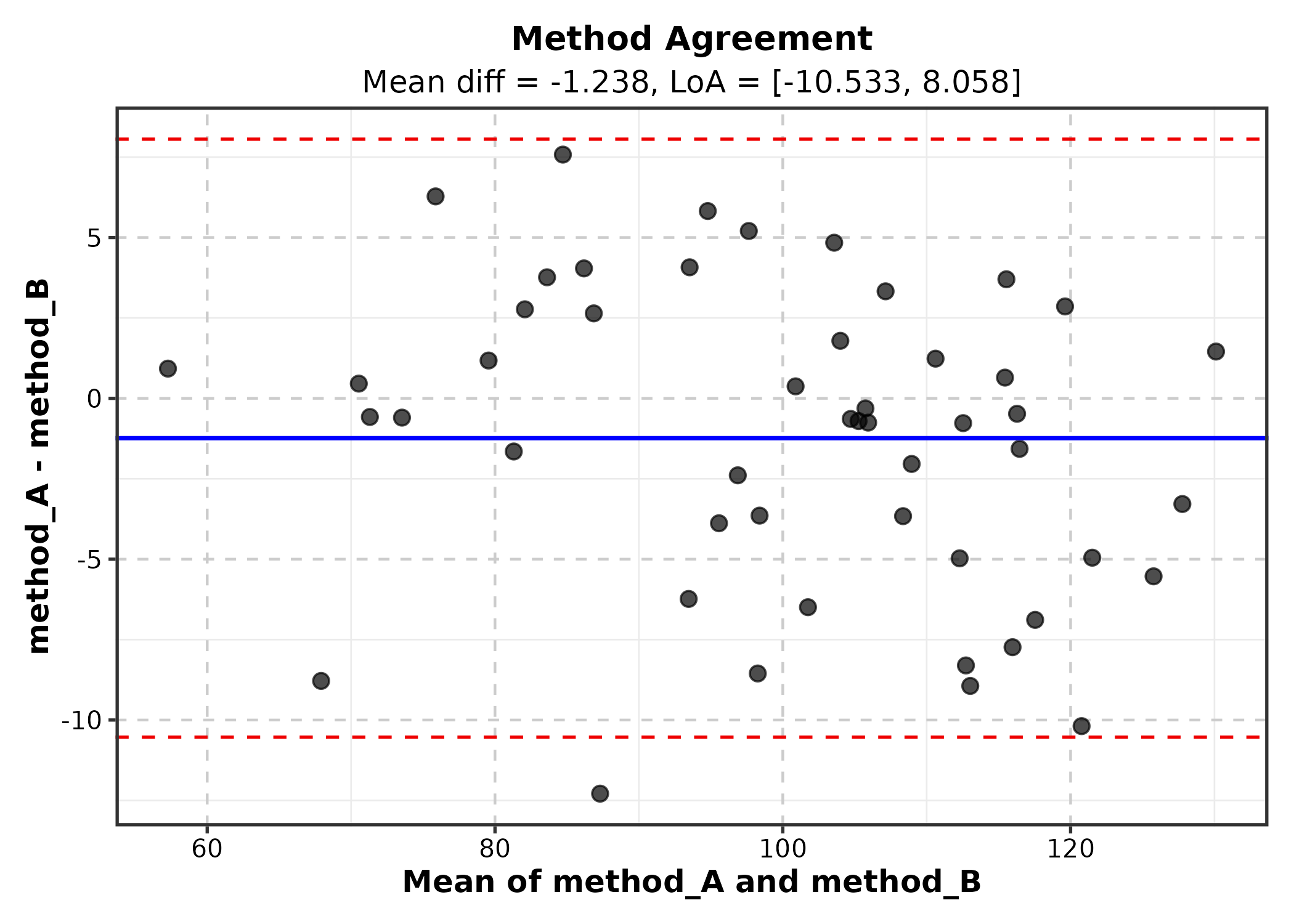
10. Ecology & Evolution
OrdinationPlot
Requires pre-computed ordination coordinates (e.g., from
vegan::metaMDS).
library(vegan)
ord <- metaMDS(species_mat, k = 2, trace = 0)
ord_df <- as.data.frame(scores(ord, display = "sites"))
ord_df$habitat <- env$habitat
OrdinationPlot(ord_df, x = "NMDS1", y = "NMDS2",
group_by = "habitat", palette = "Set1",
title = "NMDS Ordination")PhyloTreePlot
tree <- ape::rtree(12, tip.label = paste0("Species_", LETTERS[1:12]))
PhyloTreePlot(tree, palette = "Set2",
title = "Phylogenetic Tree")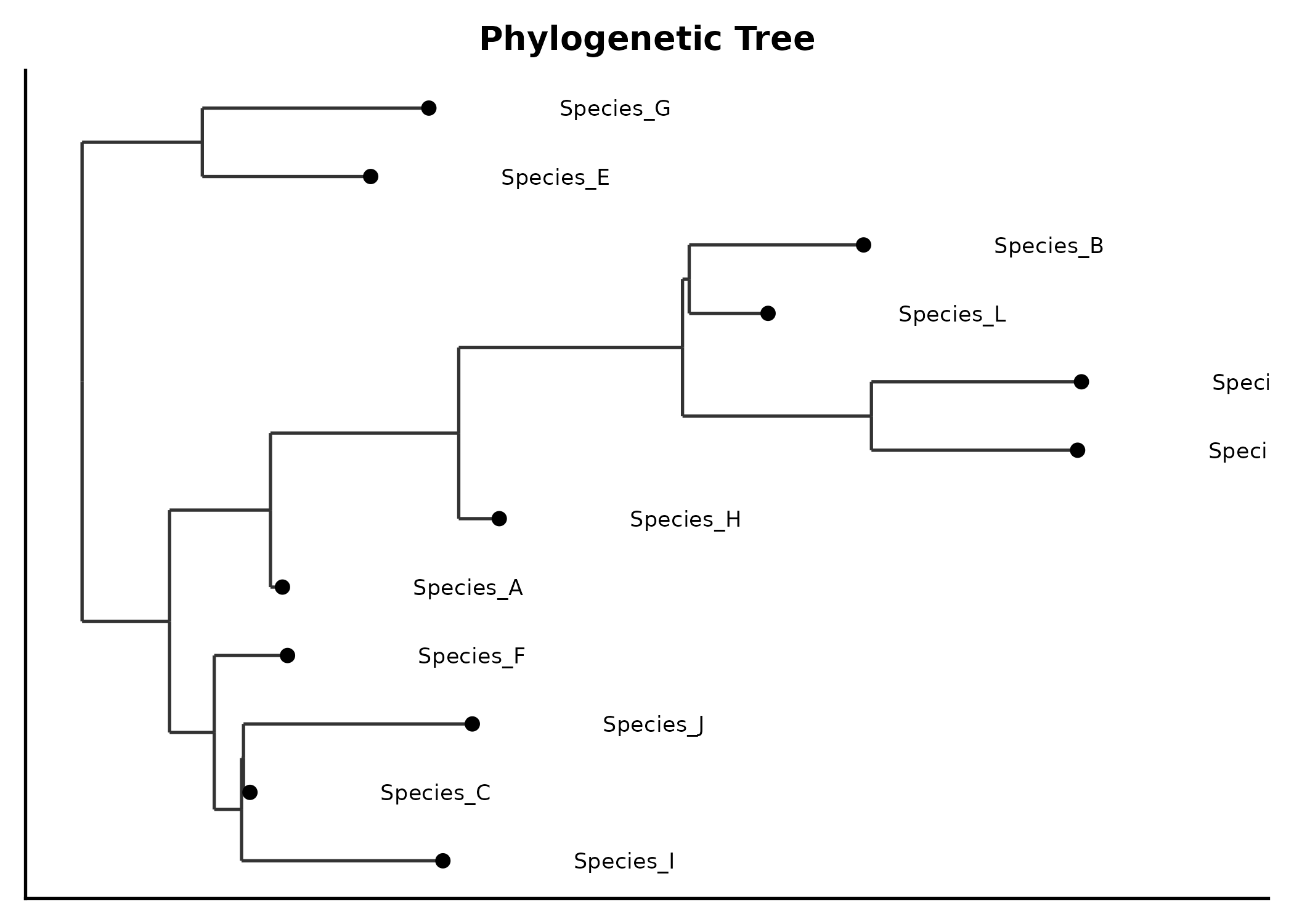
RankAbundancePlot
species = species names, abundance =
abundance values.
rank_df <- data.frame(
species = paste0("Sp", 1:30),
abundance = sort(rpois(30, 20), decreasing = TRUE),
community = sample(c("Site A", "Site B"), 30, replace = TRUE)
)
RankAbundancePlot(rank_df, species = "species", abundance = "abundance",
group_by = "community", palette = "Set1",
title = "Rank-Abundance Curve")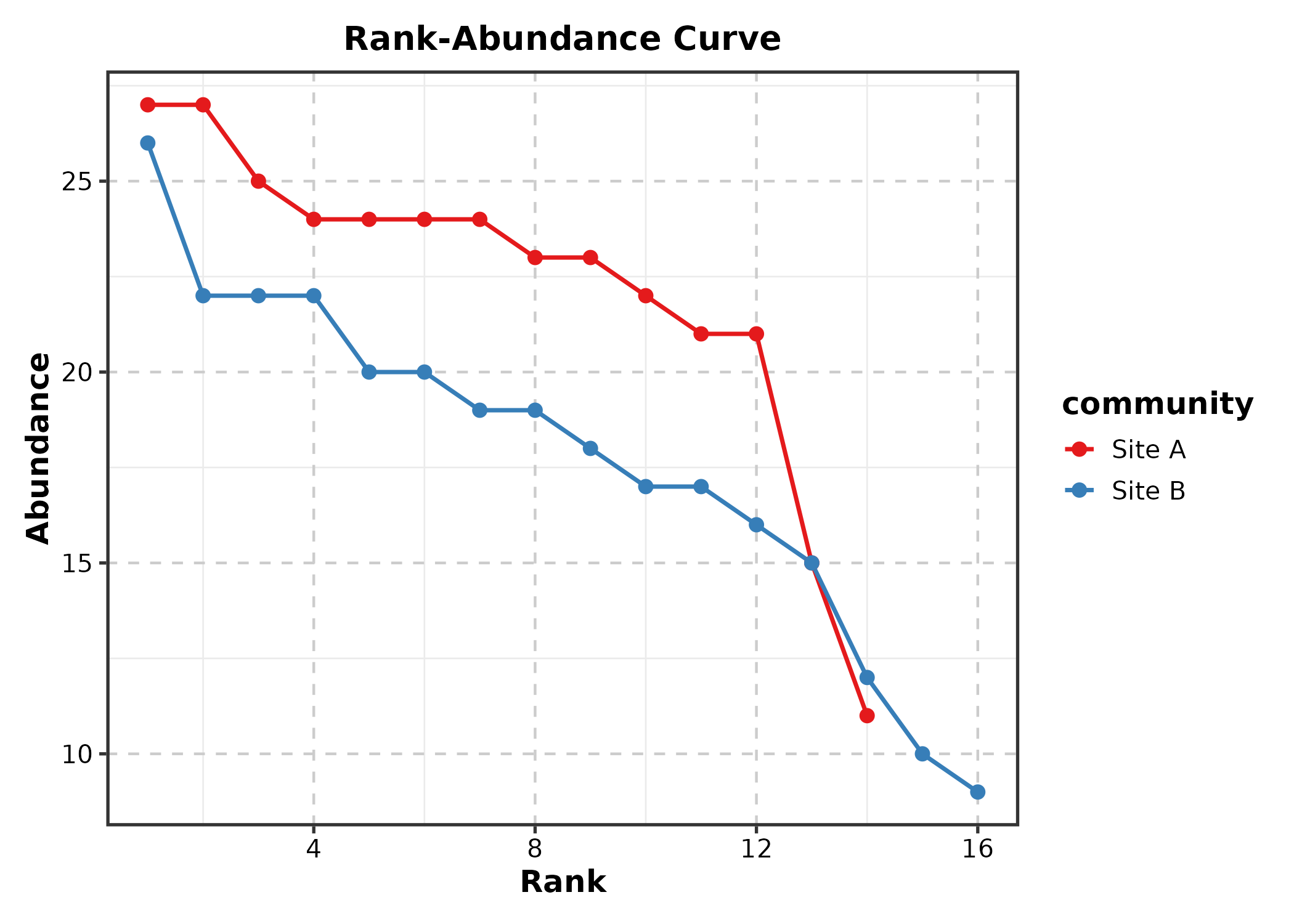
11. Physics & Engineering
QuiverPlot
field <- expand.grid(x = seq(-2, 2, 0.4), y = seq(-2, 2, 0.4))
field$u <- -field$y
field$v <- field$x
QuiverPlot(field, x = "x", y = "y", u = "u", v = "v",
title = "Vector Field")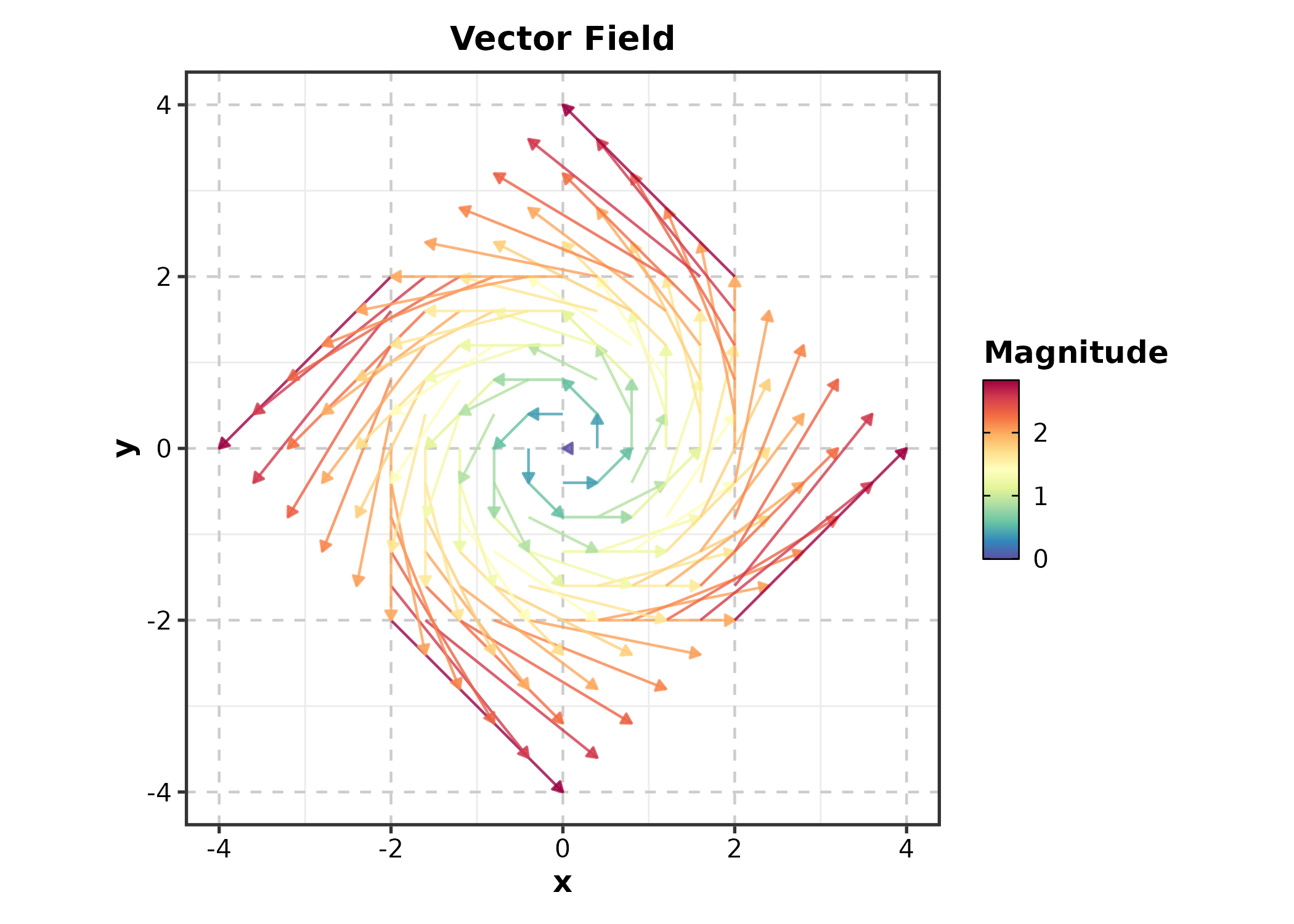
StreamlinePlot
StreamlinePlot(field, x = "x", y = "y", u = "u", v = "v",
title = "Flow Streamlines")
12. 3D & Interactive
ggforge_interactive
Converts any ggplot to an interactive plotly widget.
p <- BoxPlot(treat, x = "group", y = "response", palette = "lancet")
ggforge_interactive(p)13. Machine Learning
ConfusionMatrixPlot
truth = actual labels, predicted =
predicted labels.
set.seed(42)
pred_df <- data.frame(
actual = sample(c("Benign", "Malignant", "Normal"), 300,
replace = TRUE, prob = c(0.4, 0.35, 0.25)),
predicted = sample(c("Benign", "Malignant", "Normal"), 300,
replace = TRUE, prob = c(0.38, 0.37, 0.25))
)
diag_idx <- sample(300, 200)
pred_df$predicted[diag_idx] <- pred_df$actual[diag_idx]
ConfusionMatrixPlot(pred_df, truth = "actual", predicted = "predicted",
palette = "Blues", title = "Classification Results")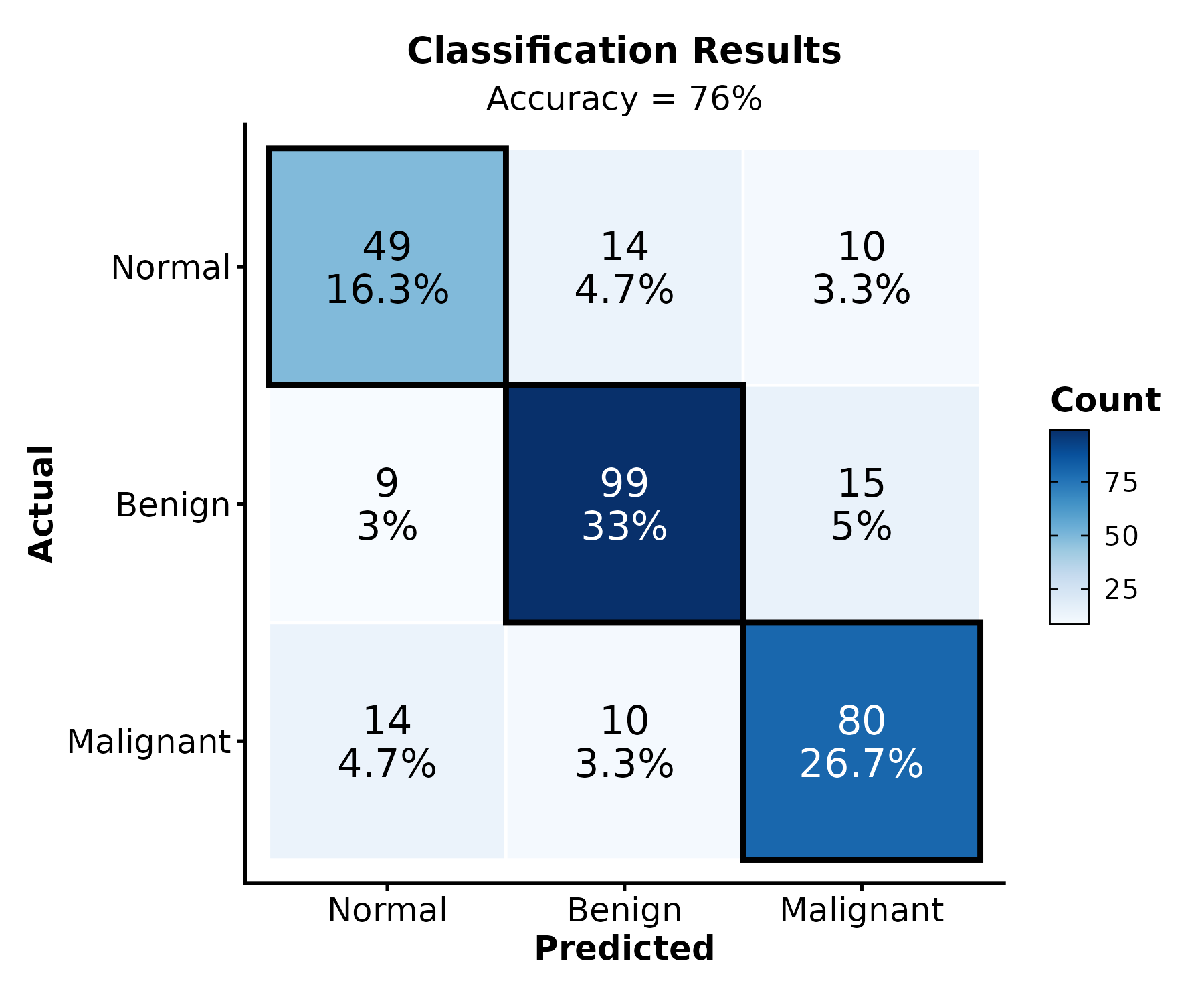
ConfusionMatrixPlot(pred_df, truth = "actual", predicted = "predicted",
normalize = "row", palette = "Greens",
title = "Row-Normalized")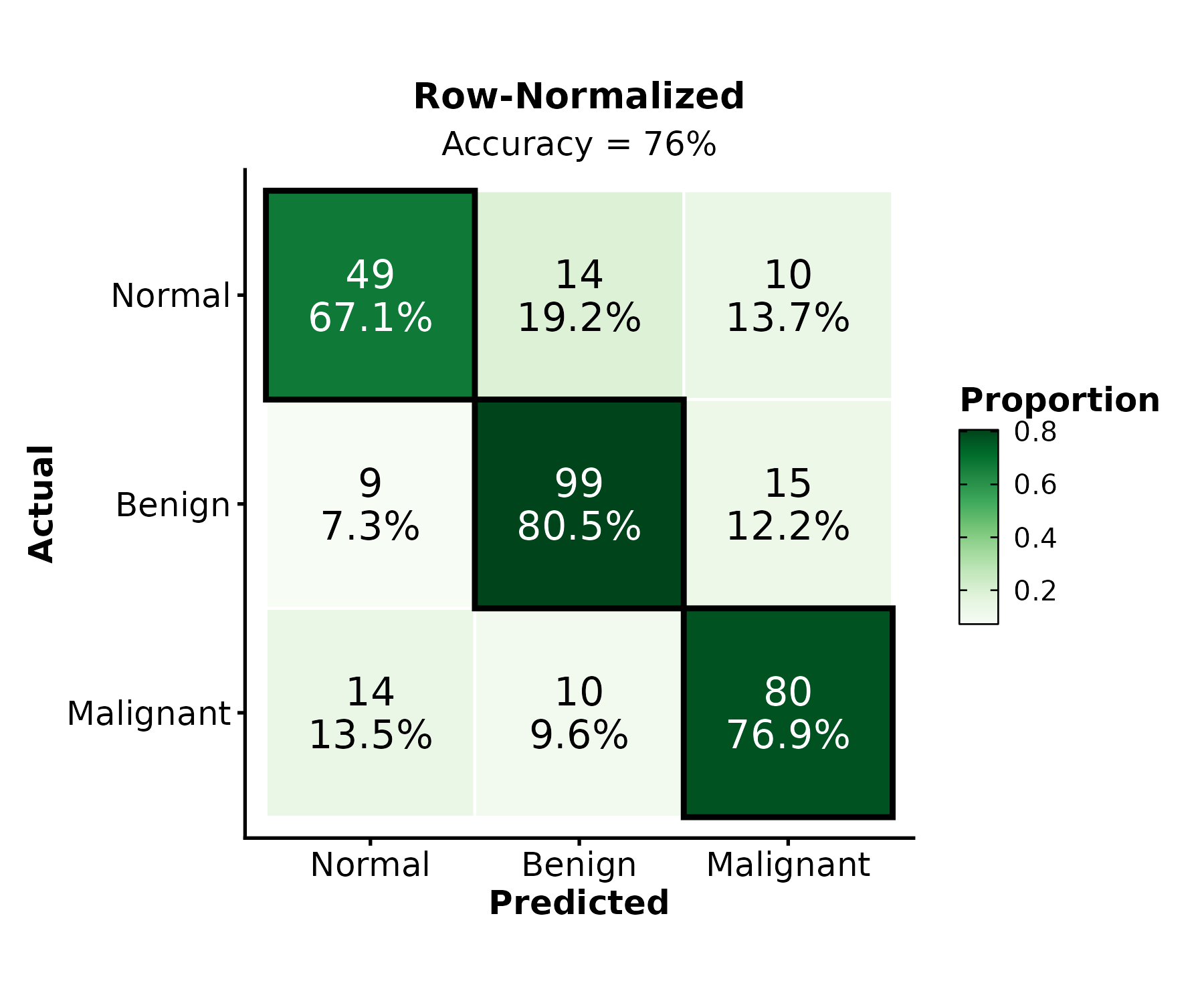
Session Info
sessionInfo()
#> R version 4.6.0 (2026-04-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] dplyr_1.2.1 ggforge_2.0.1 ggplot2_4.0.3
#>
#> loaded via a namespace (and not attached):
#> [1] RColorBrewer_1.1-3 ggdendro_0.2.0 jsonlite_2.0.0
#> [4] shape_1.4.6.1 magrittr_2.0.5 magick_2.9.1
#> [7] ggbeeswarm_0.7.3 farver_2.1.2 rmarkdown_2.31
#> [10] GlobalOptions_0.1.4 fs_2.1.0 ragg_1.5.2
#> [13] vctrs_0.7.3 Cairo_1.7-0 memoise_2.0.1
#> [16] rstatix_0.7.3 htmltools_0.5.9 forcats_1.0.1
#> [19] broom_1.0.13 gridGraphics_0.5-1 Formula_1.2-5
#> [22] sass_0.4.10 pracma_2.4.6 bslib_0.11.0
#> [25] htmlwidgets_1.6.4 desc_1.4.3 plyr_1.8.9
#> [28] zoo_1.8-15 cachem_1.1.0 ggfittext_0.10.3
#> [31] commonmark_2.0.0 igraph_2.3.2 lifecycle_1.0.5
#> [34] iterators_1.0.14 pkgconfig_2.0.3 Matrix_1.7-5
#> [37] R6_2.6.1 fastmap_1.2.0 clue_0.3-68
#> [40] digest_0.6.39 colorspace_2.1-2 ggnewscale_0.5.2
#> [43] patchwork_1.3.2 S4Vectors_0.50.1 metR_0.18.3
#> [46] textshaping_1.0.5 ggpubr_0.6.3 labeling_0.4.3
#> [49] mgcv_1.9-4 polyclip_1.10-7 abind_1.4-8
#> [52] compiler_4.6.0 withr_3.0.2 doParallel_1.0.17
#> [55] S7_0.2.2 backports_1.5.1 carData_3.0-6
#> [58] viridis_0.6.5 ggupset_0.4.1 ggforce_0.5.0
#> [61] ggsignif_0.6.4 MASS_7.3-65 proxyC_0.5.2
#> [64] rjson_0.2.23 caTools_1.18.3 tools_4.6.0
#> [67] vipor_0.4.7 otel_0.2.0 beeswarm_0.4.0
#> [70] ape_5.8-1 qqconf_1.3.2 glue_1.8.1
#> [73] treemapify_2.6.0 nlme_3.1-169 gridtext_0.1.6
#> [76] grid_4.6.0 checkmate_2.3.4 cluster_2.1.8.2
#> [79] generics_0.1.4 isoband_0.3.0 qqplotr_0.0.7
#> [82] gtable_0.3.6 tidyr_1.3.2 data.table_1.18.4
#> [85] ggVennDiagram_1.5.7 tidygraph_1.3.1 xml2_1.5.2
#> [88] car_3.1-5 BiocGenerics_0.58.1 markdown_2.0
#> [91] ggrepel_0.9.8 foreach_1.5.2 pillar_1.11.1
#> [94] stringr_1.6.0 robustbase_0.99-7 circlize_0.4.18
#> [97] splines_4.6.0 tweenr_2.0.3 lattice_0.22-9
#> [100] survival_3.8-6 tidyselect_1.2.1 ComplexHeatmap_2.28.0
#> [103] knitr_1.51 gridExtra_2.3 litedown_0.9
#> [106] IRanges_2.46.0 svglite_2.2.2 stats4_4.6.0
#> [109] ggmanh_1.16.0 xfun_0.58 graphlayouts_1.2.3
#> [112] plotROC_2.3.3 matrixStats_1.5.0 DEoptimR_1.2-0
#> [115] stringi_1.8.7 yaml_2.3.12 evaluate_1.0.5
#> [118] codetools_0.2-20 ggwordcloud_0.6.2 ggraph_2.2.2
#> [121] twosamples_2.0.1 tibble_3.3.1 cli_3.6.6
#> [124] pbmcapply_1.5.1 systemfonts_1.3.2 jquerylib_0.1.4
#> [127] dichromat_2.0-0.1 Rcpp_1.1.1-1.1 png_0.1-9
#> [130] parallel_4.6.0 ggstream_0.1.0 pkgdown_2.2.0
#> [133] ggalluvial_0.12.6 opdisDownsampling_1.0.1 bitops_1.0-9
#> [136] viridisLite_0.4.3 ggridges_0.5.7 scales_1.4.0
#> [139] purrr_1.2.2 crayon_1.5.3 GetoptLong_1.1.1
#> [142] rlang_1.2.0